Read structure
The input reads usually contain the biological sequence and optionally a cell barcode, UMI, and/or hashtag. Their location on the reads is configured in the Read structure dialog (figure 6.1). See Barcodes from names if the cell barcode is not part of the input reads.
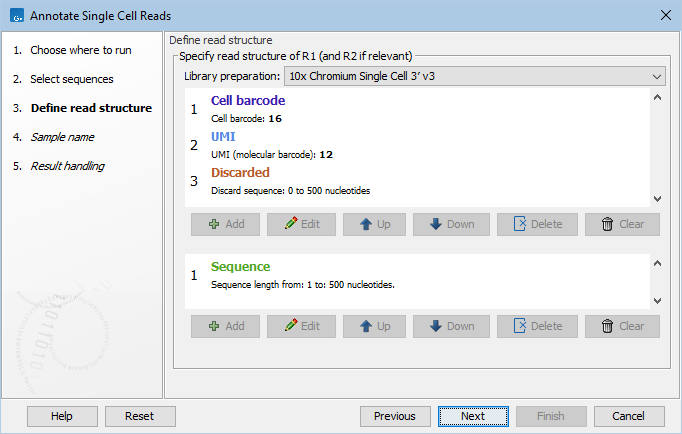
Figure 6.1: The read structure for a 10x 3' gene expression protocol.
Under Library preparation, predefined read structures are available for several protocols.
Multiome ATAC
When Library preparation is set to '10x Chromium Single Cell Multiome ATAC', the 10x Multiome ATAC barcodes are translated to 10x Multiome GEX barcodes.
This enables the combined analysis of ATAC and GEX data, for example in dimensionality reduction plots. This is illustrated in the template workflow Chromatin Accessibility and Expression Analysis from Reads. Reads with barcodes that cannot be translated are discarded.
It is not possible to customize this read structure whilst retaining the barcode translation.
Parse Biosciences
When Library preparation is set to one of the Parse Biosciences options, the barcodes are translated to the well number they originate from.
The barcode order on the read structure is reversed compared to the barcoding rounds. Thus, the first barcode on R2 corresponds to the third barcoding round. During barcode translation, the barcode order is reversed so that it corresponds to the order of the barcoding rounds.
It is not possible to customize the Parse Biosciences read structures whilst retaining the barcode translation.
After the third barcoding round, cells/nuclei are pooled and optionally further split into distinct populations, known as sublibraries. Each sublibrary has a unique fourth barcode (the Illumina sample index). When sublibraries are used, it is crucial to check one of the options from Barcodes from names to correctly annotate the reads with the fourth barcode.
Custom read structure
When Library preparation is set to 'Custom', the read structure of the protocol previously selected can be updated in the two panels below.
The top panel contains the structure of R1 of a pair, or single-end reads. The bottom panel describes R2. For single-end reads, the configuration in the bottom panel is ignored.
Five different types of tags can be used for defining the read structure:
- Sequence
- Cell barcode
- UMI
- Hashtag
- Discarded
Only the Sequence part of the read will be retained in the 'annotated reads' list. The parts of the reads corresponding to the other tags are removed. Cell barcode, UMI and Hashtag are added as annotations on the read to be used by downstream tools.
Consider the read structure from figure 6.1. R1 consists of a 16 nt cell barcode, followed by a 12 nt UMI, with an additional sequence of variable length (0 to 500 nt) that is discarded. Read pairs with an R1 shorter than 28 (=16+12) nt or longer than 528 (=16+12+500) nt do not match the read structure and will be discarded. As only R2 contains a 'Sequence' tag, the output will be single-end reads containing R2 from the original pairs, and annotated with the cell barcode and UMI from R1.
It is important that the read structure describes the full length of the read. If the variable length 'Discarded' tag was not included in R1 (figure 6.1), only read pairs with an R1 of exactly 28 nt would match the read structure.
Many different library structures are listed at https://teichlab.github.io/scg_lib_structs/. Figure 6.2 shows the configuration for Microwell-seq, as described in the resource at the time of writing: R1 contains 6 nt cell barcode + 15 nt adapter + 6 nt cell barcode + 15 nt adapter + 6 nt cell barcode + 6 nt UMI + polyA, while R2 contains the biological insert. The tool would construct a single 18 nt cell barcode from the three tags of 6nt each. More general constructions are also possible. For example, if two UMI tags are defined, one on R1 and one on R2, then a single UMI will be constructed from both parts.

Figure 6.2: Configuration for R1 from Microwell-seq. A single 18 nt cell barcode will be constructed from the three tags of 6nt each.