Quality Control of ChIP-Seq data
During the first step of the analysis, the Transcription Factor ChIP-Seq tool analyzes the input to
check if the input data satisfy the assumptions made by the algorithm
and to compute several quality measures.
The cross-correlation between reads mapping in the forward and in the reverse
strand is often used to investigate the quality of ChIP-Seq experiments
[Landt et al., 2012,Marinov et al., 2014]. The quality is determined with respect to the two main peaks of the cross-correlation plot (figure 29.2),
the peak at the read length (often called a phantom peak [Landt et al., 2012]) and the one at the fragment length.
The peak at the fragment length is typically higher than the peak at the read
length and the background (figure 29.2) for successful ChIP-Seq experiments.
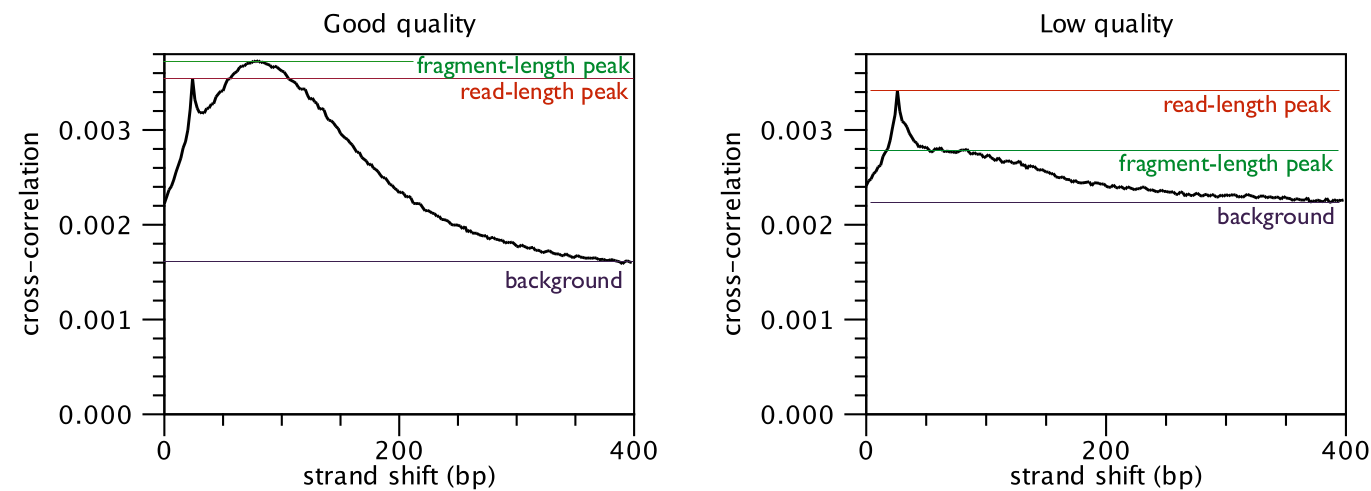
Figure 29.2: Difference in cross-correlation profiles in ChIP experiments of good and low quality.
For each input file of the analysis, the Transcription Factor ChIP-Seq tool calculates and reports several quality measures. Those quality measures have been investigated by the modENCODE consortium and are described in more detail in [Landt et al., 2012]. The quality measures are:
- Number of mapped reads
- For mammalian cells (e.g. human and mouse), this value should be at least 10 million reads. For smaller organisms such as worm and fly, this value should be at least 2 million reads.
- Normalized strand coefficient
- The normalized strand coefficient describes the ratio between the fragment-length peak and the background cross-correlation values. This value should be greater than 1.05 for ChIP-Seq experiments.
- Relative strand correlation
- The relative strand correlation describes the ratio between the fragment-length peak and the read-length peak in the cross-correlation plot. This value should be high (at least 0.8) for transcription factor binding sites, which have a concentrated signal. However, this value can be low even for successful ChIP-Seq experiments on histone modifications [Landt et al., 2012].