Alignment based design of PCR primers
In this mode, a single or a
pair of PCR primers are designed. CLC Genomics Workbench allows the user to
design primers which will specifically amplify a group of
included sequences but not amplify the remainder of the
sequences, the excluded sequences. The selection boxes are
used to indicate the status of a sequence, if the box is checked the
sequence belongs to the included sequences, if not, it belongs to
the excluded sequences. To design primers that are general for all
primers in an alignment, simply add them all to the set of included
sequences by checking all selection boxes. Specificity of priming is
determined by criteria set by the user in the dialog box which is
shown when the Calculate button is pressed (see below).
Different options can be chosen concerning the match of the primer to the template sequences in the included group:
- Perfect match. Specifies that the designed primers must have a perfect match to all relevant sequences in the alignment. When selected, primers will thus only be located in regions that are completely conserved within the sequences belonging to the included group.
- Allow degeneracy. Designs primers that may include ambiguity characters
where heterogeneities occur in the included template sequences. The
allowed fold of degeneracy is user defined and corresponds to the number of possible primer combinations formed
by a degenerate primer. Thus, if a primer covers two 4-fold degenerate site and one 2-fold degenerate site
the total fold of degeneracy is
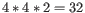 and the primer will, when supplied from the manufacturer, consist of a
mixture of 32 different oligonucleotides. When scoring the available primers, degenerate primers are given a score which decreases
with the fold of degeneracy.
and the primer will, when supplied from the manufacturer, consist of a
mixture of 32 different oligonucleotides. When scoring the available primers, degenerate primers are given a score which decreases
with the fold of degeneracy.
- Allow mismatches. Designs primers which are allowed a
specified number of mismatches to the included template sequences. The melting temperature algorithm employed includes the latest thermodynamic
parameters for calculating
 when single-base mismatches occur.
when single-base mismatches occur.
When in Standard PCR mode, clicking the Calculate button will prompt the dialog shown in figure 16.14.
The top part of this dialog shows the single-primer parameter settings chosen in the Primer parameters preference group which will be used by the design algorithm.
The central part of the dialog contains parameters pertaining to primer specificity (this is omitted if all sequences belong to the included group). Here, three parameters can be set:
- Minimum number of mismatches - the minimum number of mismatches that a primer must have against all sequences in the excluded group to ensure that it does not prime these.
- Minimum number of mismatches in 3' end - the minimum number of mismatches that a primer must have in its 3' end against all sequences in the excluded group to ensure that it does not prime these.
- Length of 3' end - the number of consecutive nucleotides to consider for mismatches in the 3' end of the primer.
The lower part of the dialog contains parameters pertaining to primer pairs (this is omitted when only designing a single primer). Here, three parameters can be set:
- Maximum percentage point difference in G/C content - if this is set at e.g. 5 points a pair of primers with 45% and 49% G/C nucleotides, respectively, will be allowed, whereas a pair of primers with 45% and 51% G/C nucleotides, respectively will not be included.
- Maximal difference in melting temperature of primers in a pair - the number of degrees Celsius that primers in a pair are all allowed to differ.
- Max hydrogen bonds between pairs - the maximum number of hydrogen bonds allowed between the forward and the reverse primer in a primer pair.
- Maximum length of amplicon - determines the maximum length of the PCR fragment.
The output of the design process is a table of single primers or primer pairs as described for primer design based on single sequences. These primers are specific to the included sequences in the alignment according to the criteria defined for specificity. The only novelty in the table, is that melting temperatures are displayed with both a maximum, a minimum and an average value to reflect that degenerate primers or primers with mismatches may have heterogeneous behavior on the different templates in the group of included sequences.
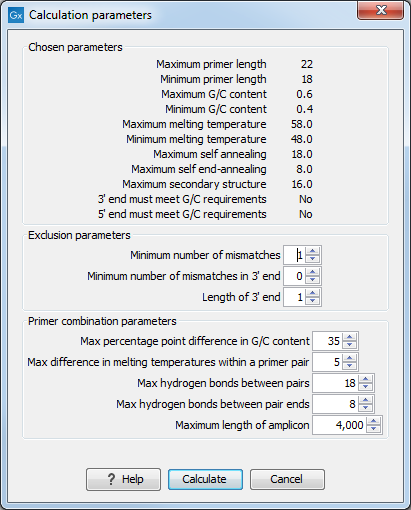
Figure 16.14: Calculation dialog shown when designing alignment based PCR primers.