Protein coloring to visualize local structural uncertainties
The default coloring scheme for modeled structures in CLC Genomics Workbench is "Color by Temperature". This coloring indicates the uncertainty or disorder of each atom position in the structure.
For crystal structures, the temperature factor (also called the B-factor) is given in the PDB file as a measure of the uncertainty or disorder of each atom position. The temperature factor has the unit Å2, and is typically in the range [0, 100].
The temperature color scale ranges from blue (0) over white (50) to red (100) (see Visualization styles and colors).
For structure models created in CLC Genomics Workbench, the temperature factor assigned to each atom combines three sources of positional uncertainty:
- PDB Temp. The atom position uncertainty for the template structure, represented by the temperature factor of the backbone atoms in the template structure.
- P(alignment) The probability that the alignment of a residue in the query sequence to a particular position on the structure is correct.
- Clash? It is evaluated if atoms in the structure model seem to clash, thereby indicating a problem with the model.
The three aspects are combined to give a temperature value between zero and 100, as illustrated in figure 25.82 and 25.83.
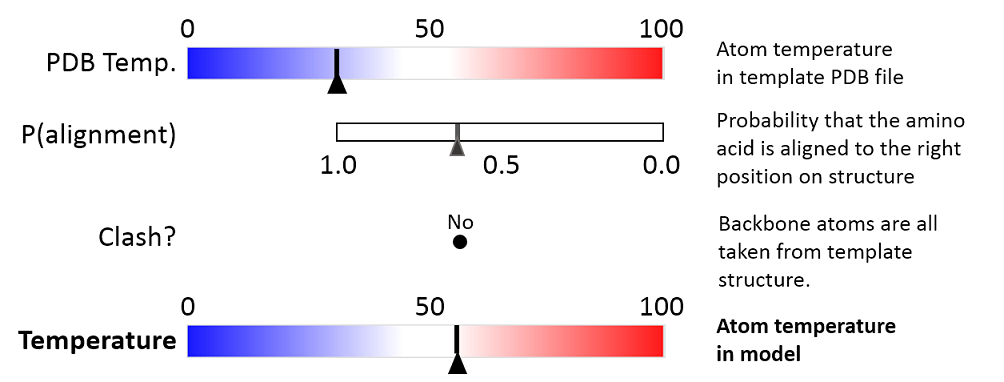
Figure 25.82: Evaluation of temperature color for backbone atoms in structure models.
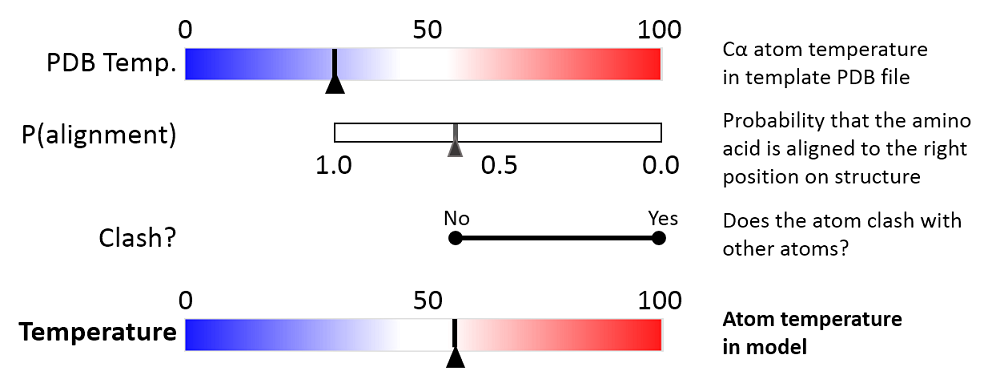
Figure 25.83: Evaluation of temperature color for side chain atoms in structure models.
When holding the mouse over an atom, the Property Viewer in the Side Panel will show various information about the atom. For atoms in structure models, the contributions to the assigned temperature are listed as seen in figure 25.84.
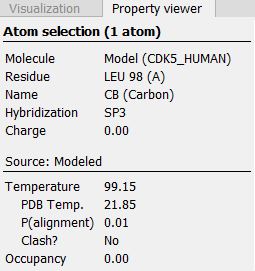
Figure 25.84: Information displayed in the Side Panel Property viewer for a modeled atom.
|
Note: For NMR structures, the temperature factor is set to zero in the PDB file, and the "Color by Temperature" will therefore suggest that the structure is more well determined than is actually the case. |
P(alignment)
Alignment error is one of the largest causes of model inaccuracy, particularly when the model is built from a template sharing low sequence identity (e.g. lower than 60%). Misaligning a single amino acid by one position will cause a ca. 3.5 Å shift of its atoms from their true positions.
The estimate of the probability that two amino acids are correctly aligned, P(alignment), is obtained by averaging over all the possible alignments between two sequences, similar to [Knudsen and Miyamoto, 2003].
This allows local alignment uncertainty to be detected even in similar sequences. For example the position of the D in this alignment:
| Template | GGACDAEDRSTRSTACE---GG |
| Target | GGACD---RSTRSTACEKLMGG |
is uncertain, because an alternative alignment is as likely:
| Template | GGACDAEDRSTRSTACE---GG |
| Target | GGAC---DRSTRSTACEKLMGG |
Clash?
Clashes are evaluated separately for each atom in a side chain. The scoring function used to evaluate protein-ligand interactions for docking and ligand optimization in the now discontinued CLC Drug Discovery Workbench is also used to evaluate the interaction between a given side chain atom and its surroundings.
If the atom is considered to clash, it will be assigned a temperature of 100.
|
Note: Clashes within the modeled protein chain as well as with all other molecules in the downloaded PDB file (except water) are considered. |