Trimming using the Trim tool
Sequence reads can be trimmed based on a number of
different criteria. Using a trimming tool for this is particularly useful if:
- You have many sequences to trim.
- You wish to trim vector contamination from sequencing reads.
- You wish to ensure that consistency when trimming. That is, you wish to ensure the same criteria are used for all the sequences in a set.
To start up the Trim tool in the Workbench, go to the menu option:
Toolbox | Molecular Biology Tools (![]() ) | Sanger Sequencing Analysis (
) | Sanger Sequencing Analysis (![]() )| Trim Sequences (
)| Trim Sequences (![]() )
)
This opens a dialog where you can choose the sequences to trim, by using the arrows to move them between the Navigation Area and the 'Selected Elements' box.
You can then specify the trim parameters as displayed in figure 18.5.
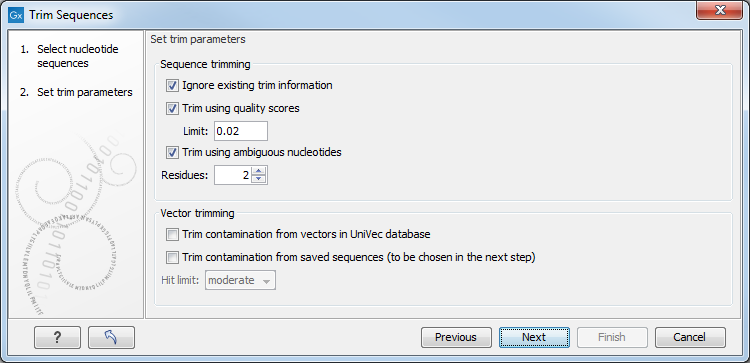
Figure 18.5: Setting parameters for trimming.
The following parameters can be adjusted in the dialog:
- Ignore existing trim information. If you have previously trimmed the sequences, you can check this to remove existing trimming annotation prior to analysis.
- Trim using quality scores. If the sequence files contain quality
scores from a base caller algorithm this information can be used for
trimming sequence ends. The program uses the modified-Mott trimming
algorithm for this purpose (Richard Mott, personal communication):
Quality scores in the Workbench are on a Phred scale, and formats using other scales will be converted during import. The Phred quality scores (Q), defined as:
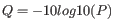 , where P is the base-calling error probability, can then be used to calculate the error probabilities, which in turn can be used to set the limit for, which bases should be trimmed.
, where P is the base-calling error probability, can then be used to calculate the error probabilities, which in turn can be used to set the limit for, which bases should be trimmed.
Hence, the first step in the trim process is to convert the quality score (Q) to an error probability:
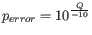 . (This now means that low values are high quality bases.)
. (This now means that low values are high quality bases.)
Next, for every base a new value is calculated:
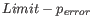 . This value will be negative for low quality bases, where the error probability is high.
. This value will be negative for low quality bases, where the error probability is high.
For every base, the Workbench calculates the running sum of this value. If the sum drops below zero, it is set to zero. The part of the sequence not trimmed will be the region ending at the highest value of the running sum and starting at the last zero value before this highest score. Everything before and after this region will be trimmed. A read will be completely removed if the score never makes it above zero.
- Trim ambiguous nucleotides. This option trims the sequence ends based on the presence of ambiguous nucleotides (typically N). Note that the automated sequencer generating the data must be set to output ambiguous nucleotides in order for this option to apply. The algorithm takes as input the maximal number of ambiguous nucleotides allowed in the sequence after trimming. If this maximum is set to e.g. 3, the algorithm finds the maximum length region containing 3 or fewer ambiguities and then trims away the ends not included in this region. The "Trim ambiguous nucleotides" option trims all types of ambiguous nucleotides (see IUPAC codes for nucleotides).
- Trim contamination from vectors in UniVec database. If selected, the program will match the sequence reads against all vectors in the UniVec database and mark sequence ends with significant matches with a 'Trim' annotation (the database is
included when you install the CLC Genomics Workbench). A list of all the
vectors in the UniVec database can be found at
http://www.ncbi.nlm.nih.gov/VecScreen/replist.html.
- Hit limit. Specifies how strictly vector contamination is trimmed.
Since vector contamination usually occurs at the beginning or end of
a sequence, different criteria are applied for terminal and internal
matches. A match is considered terminal if it is located within the
first 25 bases at either sequence end. Three match categories are
defined according to the expected frequency of an alignment with the
same score occurring between random sequences. The CLC Genomics Workbench uses the same settings as VecScreen (http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html):
- Weak. Expect 1 random match in 40 queries of length 350 kb
- Terminal match with Score 16 to 18.
- Internal match with Score 23 to 24.
- Moderate. Expect 1 random match in 1,000 queries of length 350 kb
- Terminal match with Score 19 to 23.
- Internal match with Score 25 to 29.
- Strong. Expect 1 random match in 1,000,000 queries of length 350 kb
- Terminal match with Score
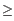 24.
24.
- Internal match with Score 30.
- Terminal match with Score
- Weak. Expect 1 random match in 40 queries of length 350 kb
- Hit limit. Specifies how strictly vector contamination is trimmed.
Since vector contamination usually occurs at the beginning or end of
a sequence, different criteria are applied for terminal and internal
matches. A match is considered terminal if it is located within the
first 25 bases at either sequence end. Three match categories are
defined according to the expected frequency of an alignment with the
same score occurring between random sequences. The CLC Genomics Workbench uses the same settings as VecScreen (http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html):
- Trim contamination from saved sequences. This option lets you select your own vector sequences that you have imported into the Workbench. If you select this option, you will be able to select one or more sequences when you click Next.
Click Finish to start the tool. This will start the trimming process. Views of each trimmed sequence will be shown, andyou can inspect the result by looking at the "Trim" annotations (they are colored red as default). Note that the trim annotations are used to signal that this part of the sequence is to be ignored during further analyses, hence the trimmed sequences are not deleted. If there are no trim annotations, the sequence has not been trimmed.