Extract Annotations
The Extract annotations tool makes it very easy to extract parts of a sequence (or several sequences) based on its annotations. In just a few steps, it becomes possible to:
- extract all tRNA genes from a genome.
- automatically add flanking regions to the annotated sequences.
- search for specific words in all available annotations.
- extract nucleotide sequences of differentially expressed genes or transcripts when using RNA-seq statistical comparisons as input.
The output is a sequence list that contains sequences carrying the annotation specified (including the flanking regions, if this option was selected).
To extract annotations from a sequence, go to:
Toolbox | Utility Tools | Extract Annotations (![]() )
)
This opens the dialog shown in figure 32.9 where you can select one or more annotated sequences, or annotation tracks, or variant tracks, or statistical comparisons. Click Next.
Figure 32.9: Select one or more annotated sequences, annotations, variant tracks or (in this figure) statistical comparisons.
If you selected tracks as input, the next step will ask for a reference sequence track to use for extracting the annotations, or which annotations to use if an annotated sequence was selected as input (figure 32.10).
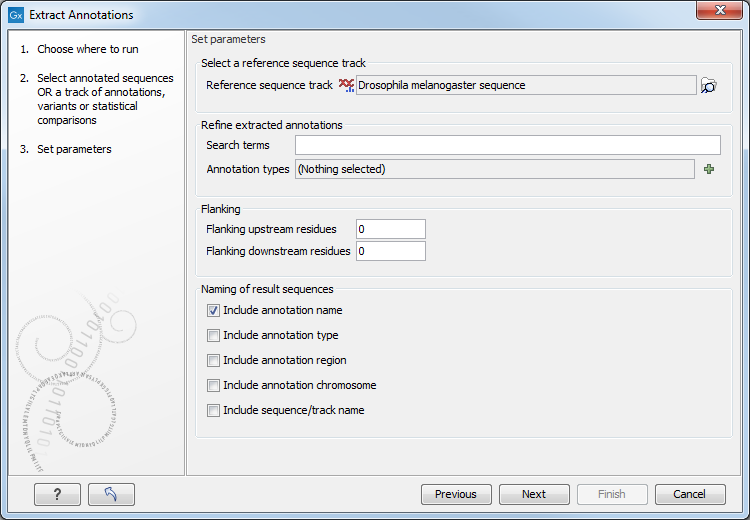
Figure 32.10: Adjusting parameters for extract annotations.
- Search terms. All annotations and attached information for each annotation will be searched for the entered term. It can be used to make general searches for search terms such as "Gene" or "Exon", or it can be used to make more specific searches. For example, if you have a gene annotation called "MLH1" and another called "MLH3", you can extract both annotations by entering "MLH" in the search term field. If you wish to enter more specific search terms, separate them with commas: "MLH1, Human" will find annotations including both "MLH1" and "Human".
- Annotation types If only certain types of annotations should be extracted, this can be specified here.
The sequence of interest can be extracted with flanking sequences:
- Flanking upstream residues. The output will include this number of extra residues at the 5' end of the annotation.
- Flanking downstream residues. The output will include this number of extra residues at the 3' end of the annotation.
The sequences that are created can be named after the annotation name, type etc:
- Include annotation name. This will use the name of the annotation in the name of the extracted sequence.
- Include annotation type. This corresponds to the type chosen above and will put this information in the name of the resulting sequences. This is useful information if you have chosen to extract "All" types of annotations.
- Include annotation region. The region covered by the annotation on the original sequence (i.e. not including flanking regions) will be included in the name.
- Include sequence/track name. If you have selected more than one sequence as input, this option enables you to discern the origin of the resulting sequences in the list by putting the name of the original sequence into the name of the resulting sequences.
Click Finish to start the tool.