Run the Local Realignment tool
The tool is found in the Toolbox:
Toolbox | Resequencing Analysis | Local Realignment (![]() )
)
Select one or multiple read mappings as input. If one read mapping is selected, local realignment will attempt to realign all contained reads, if appropriate. If multiple read mappings are selected, their reference genome must exactly match. Local realignment will realign all reads from all input read mappings as if they came from the same input. However, local realignment will create one output read mapping for each input read mapping, thereby preserving the affiliation of each read to its sample. Clicking Next allows you to set parameters as displayed in figure 25.36.
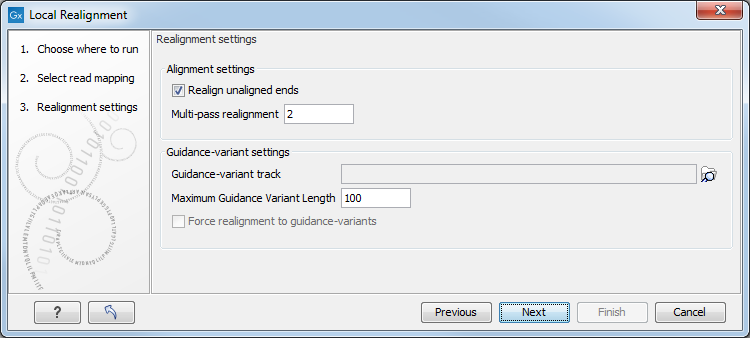
Figure 25.36: Set the realignment options.
Alignment settings
- Realign unaligned ends This option, if enabled, will trigger the realignment algorithm to attempt to realign unaligned ends as described in section "Realignment of unaligned ends (soft clipped reads)". This option should be enabled by default unless unaligned ends arise from known artifacts (such as adapter remainders in amplicon sequencing setups) and are thus not expected to be realignable anyway. Ignoring unaligned ends will yield a significant run time improvement in those cases. Realigning unaligned ends under normal conditions (where unaligned ends are expected to be realignable), however, does not contribute a lot of processing time.
- Multi-pass realignment This option is used to specify how many realignment passes should be performed by the algorithm. More passes improve accuracy at the cost of longer run time (approx. 25% per pass). Two passes are recommended; more than three passes barely yield further improvements.
Guidance-variant settings
- Guidance-variant track A track of variants to guide realignment of reads. Guiding can be used in at least two scenarios: (1) if reads are short or expected variants are long and (2) if cross sample comparisons are performed and some samples are already well genotyped. A track of variants can be produced by any of the variant detection tool, the Indels and Structural Variants tool or by importing variants from external data sources, such as dbSNP, etc.
- Maximum Guidance Variant Length set at 200 by default but can be increased to include guidance variants longer than 200 bp.
There are two modes for using the guidance track:
- Un-forced If the 'Force realignment to guidance-variants' is un-ticked the guidance variants are used as 'weak' prior evidence: each guidance variant will be represented by a pseudo-read, allowing the local realignment to explore the alignments that the guidance variants suggest. Any variant track may be used to guide the realignment when the un-forced mode is chosen.
- Force realignment to guidance-variants If the 'Force realignment to guidance-variants' is ticked the guidance variants are used as 'strong' prior evidence: a 'pseudo' reference will be generated for each guidance variant, and the alignment of nucleotides to their sequences will be awarded and encouraged as much as the alignment to the original reference sequence. Thus, the 'Force realignment to guidance-variants' options should only be used when there is prior information that the variants in the guidance variant track are infact present in the sample. This would e.g. be the case for an 'InDel' track produced by the Structural Variant tool (see Section 26.12), in an analysis of the same sample as the realignment is carried out on. Using 'forced' realignment to a general variant data base track is generally strongly discouraged.
The next dialog allows specification of the result handling. Under "Output options" it is possible to specify whether the results should be presented as a reads track or a stand-alone read mapping (figure 25.37).
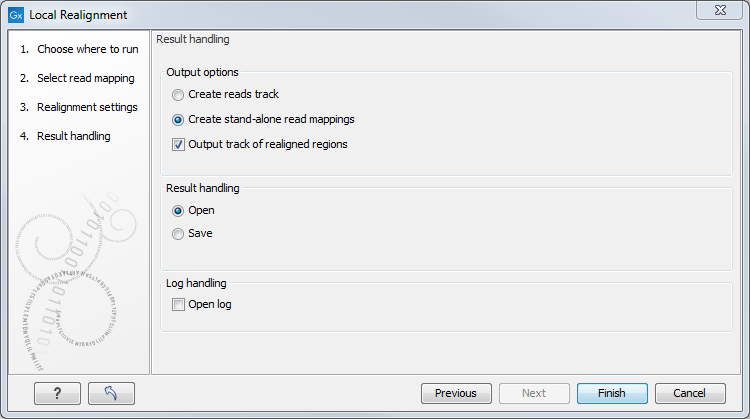
Figure 25.37: An output track of realigned regions can be created.
If enabled, the option Output track of realigned regions will cause the algorithm to output a track of regions that help pinpoint regions that have been improved by local realignment. This track has purely informative intention and cannot be used for anything else.