Remove Marginal Variants
Variant calling is always a balance between sensitivity and specificity.
To get rid of potential false positive variants, you can use this tool on
a variant track to remove some of the variant calls, which are supported by only
low quality bases, have low frequency or a skewed forward-reverse reads balance. In this way, you can
try different strategies for filtering without re-running the variant detection.
Toolbox | Resequencing Analysis | Variant Filtering | Remove Marginal Variants
This opens a dialog where you can select a variant track (![]() ) with experimental data that should be filtered.
) with experimental data that should be filtered.
Click Next to set the filtering thresholds as shown in figure 27.26
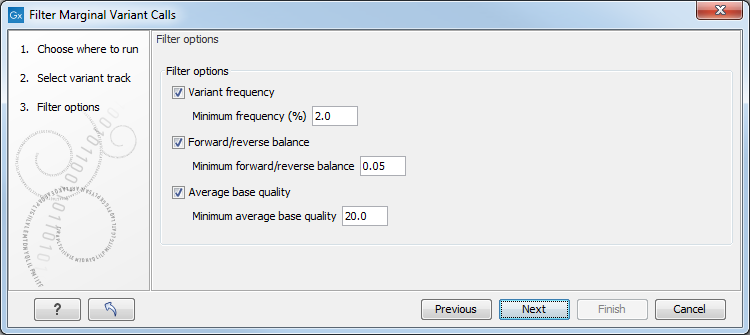
Figure 27.26: Specifying thresholds for filtering.
The following thresholds can be specified. All alleles except the reference allele are investigated separately, but in order to remove a variant, all non-reference alleles have to fulfill the requirements.
- Variant frequency
- The frequency filter will remove all variants having alleles with a frequency (= number of reads supporting the allele/number of all reads) lower than the given threshold.
- Forward/reverse balance
- The forward/reverse balance filter will remove all variants having alleles with a forward/reverse balance of less than the given threshold.
- Average base quality
- The average base quality filter will remove all variants having alleles with an average base quality of less than the given threshold.
The result is a new track where all variants (or at least one non-reference allele of the variant) fulfill the criteria.