Specifying RNA-Seq outputs
Clicking Next will allow you to specify the output options as shown in figure 28.11.
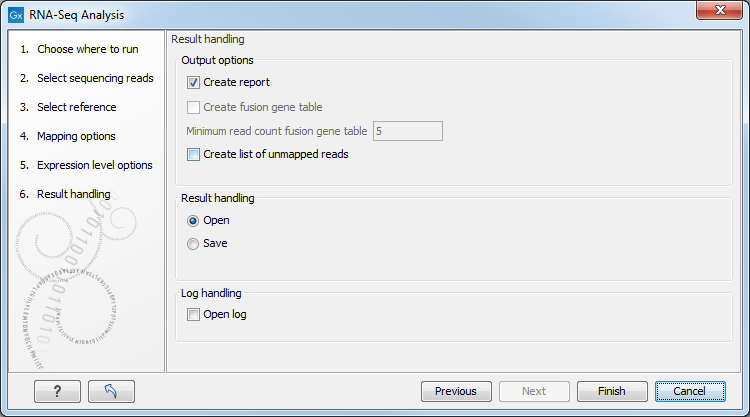
Figure 28.11: Selecting the output of the RNA-Seq analysis.
The main results of the RNA-Seq analysis are:
- Expression Tracks One track summarizing expression at the gene level is produced. The track name ends in (GE). If the "Genome annotated with genes and transcripts" option was selected, as shown in figure 28.4, then a second track summarizing expression at the transcript level is also produced. This track has a name ending with (TE).
- Reads track This track contains the mapping of the reads to the references. This track has a name ending with (Reads).
In addition, the following optional results can be selected:
- Create list of unmapped reads. Creates a list of the reads that either did not map to the reference at all or that were non-specific matches with more placements than specified (see Defining mapping options for RNA-Seq). If you started with paired reads then more than one list of unmapped reads may be produced: paired reads are put in one list, with a name that ends in (paired), singe reads, including members of broken pairs, are put in a read list with a name than ends in (single).
- Create report. Creates a report of the results. See RNA-Seq report below for a description of the information contained in the report. This report is also the only place results of the spike-in controls will be available.
- Create fusion gene table. An option that is enabled when using paired data. Creates a table that lists potential fusion genes. This, along with the Minimum read count, is described further below in section Gene fusion reporting.