Differential Expression for RNA-Seq
The Differential Expression for RNA-Seq tool performs a statistical differential expression test for a set of Expression Tracks. It uses multi-factorial statistics based on a negative binomial GLM. The tool supports paired designs and can control for batch effects. The statistical analysis is described in more detail in The statistical model.
To run the Differential Expression for RNA-Seq analysis:
Toolbox | RNA-Seq Analysis | Differential Expression for RNA-Seq
Select a number of Expression tracks (![]() ) and click Next figure 28.27.
) and click Next figure 28.27.
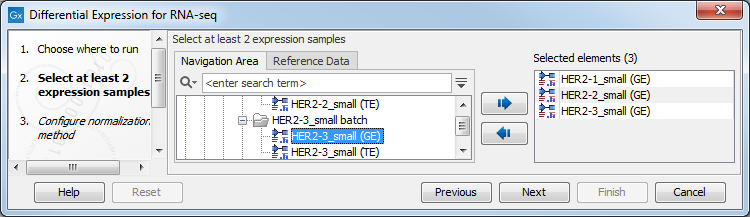
Figure 28.27: Select a number of Expression tracks.
For Expression Tracks (TE), the values used as input are "Total transcript reads". For Gene Expression Tracks (GE), the values used depend on whether an eukaryotic or prokaryotic organism is analyzed, i.e. if the option "Genome annotated with Genes and transcripts" or "Genome annotated with Genes only" is used. For Eukaryotes the values are "Total Exon Reads", whereas for Prokaryotes the values are "Total Gene Reads".
The normalization options available can be seen in figure 28.28.
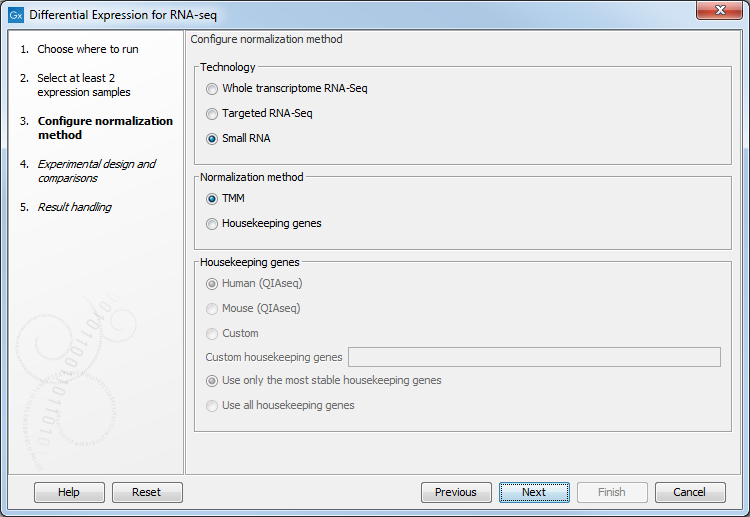
Figure 28.28: Normalization methods.
First, choose the application that was used to generate the expression tracks selected at the previous step: Whole transcriptome RNA-Seq, Targeted RNA-Seq and Small RNA. For Targeted RNA-Seq and Small RNA, you can choose between two normalization methods: TMM and Housekeeping genes, while Whole transcriptome RNA-Seq will be normalized by default using the TMM method.
TMM Normalization (Trimmed Mean of M values) calculates effective libraries sizes, which are then used as part of the per-sample normalization. TMM normalization adjusts library sizes based on the assumption that most genes are not differentially expressed.
Normalization with Housekeeping genes for can be done when a set of housekeeping genes to use is available: in the "Custom housekeeping genes" field, type the name of the genes separated by a space. Finally choose between these two options:
- Use only the most stable housekeeping genes will use a subset (at least three) of the most stable genes for normalization, these being defined using the GeNorm algorithm [Vandesompele et al., 2002].
- Use all housekeeping genes keep all housekeeping genes listed for normalization.
When working with Targeted RNA Panels, it is recommended to normalize with the Housekeeping genes method rather than TMM. Predefined list of housekeeping genes are available for samples generated using Human and Mouse QIAseq panels (hover with the mouse on the dialog to find the list of genes included in the set). If you are working with a custom panel, you can also provide the corresponding set of housekeeping genes in the "Custom housekeeping genes" as described above.
In the Experimental design panel (figure 28.29), a Metadata table must be selected that describes the factors and groups for all the samples.
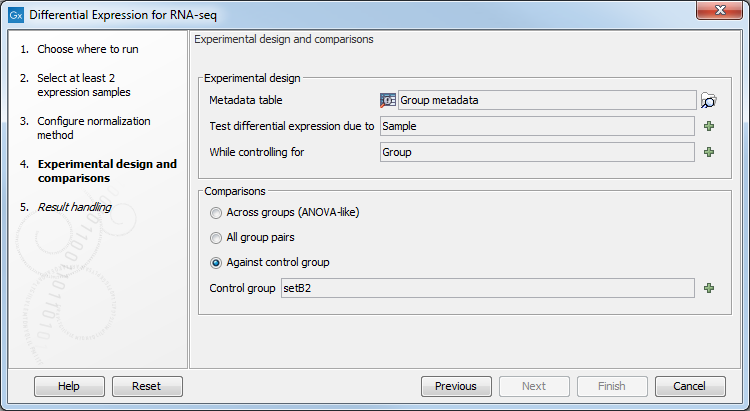
Figure 28.29: Setting up the experimental design and comparisons.
- Metadata table The metadata table describing the factors for the selected Expression tracks.
- Test differential expression due to Specify the one factor differential expression is tested for.
- While controlling for Specify confounding factors, i.e., factors that are not of primary interest, but may affect gene expression.
The Comparisons panel determines the number and type of statistical comparison tracks output by the tool (see Output of the Differential Expression for RNA-Seq tool for more details).
The Differential Expression for RNA-Seq tool produces different numbers and types of statistical comparison tracks depending on the settings of the Comparisons panel. Depending on the choice either a Wald test or a Likelihood Ratio test is used. For example, assume that we test a factor called 'Tissue' with three groups: skin, liver, brain.
- Across groups (ANOVA-like) This mode tests for the effect of a factor across all groups.
- Outputs produced: "Due to Tissue"
- Test used: Likelihood ratio test
- Fold change reports: The maximum pairwise fold change between any two of the three tissue types.
- Max of group means reports: The maximum of the average group RPKM values among any of the tissue types for a gene.
- All group pairs tests for differences between all pairs of groups in a factor.
- Outputs produced: "skin vs. liver", "skin vs. brain", "liver vs. brain"
- Test used: Wald test
- Fold change reports: The fold change in the defined order between the named pair of tissue types.
- Max of group means reports: The maximum of the average group RPKM values between the two named tissue types.
- Against control group This mode tests for differences between all the groups in a factor and the named reference group. In this example the reference group is skin.
- Outputs produced: "liver vs. skin", "brain vs. skin"
- Test used: Wald test
- Fold change reports: The fold change in the defined order between the named pair of tissue types.
- Max of group means reports: The maximum of the average group RPKM values between the two named tissue types.