Maximum likelihood reconstruction methods
Maximum likelihood (ML) based reconstruction methods [
Felsenstein, 1981] seek to identify the most probable tree given the data available, i.e. maximize
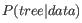
where the
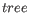
refers to a tree topology with branch lengths while
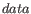
is usually a set of sequences. However, it is not possible to compute
so instead ML based methods have to compute the probability of the data given a tree, i.e.
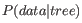
. The ML tree is then the tree which makes the data most probable. In other words, ML methods search for the tree that gives the highest probability of producing the observed sequences. This is done by searching through the space of all possible trees while computing an ML estimate for each tree. Computing an ML estimate for a tree is time consuming and since the number of tree topologies grows exponentially with the number of leaves in a tree, it is infeasible to explore all possible topologies. Consequently, ML methods must employ search heuristics that quickly converges towards a tree with a likelihood close to the real ML tree.
The likelihood of trees are computed using an explicit model of
evolution such as the Jukes-Cantor or Kimura 80 models. Choosing the right model is often important to get a good result. To help users choose the correct model for a data set, the Model Testing tool (see Model
Testing tool) can be used to test a range of different models for input nucleotide sequences.
Choosing the
right model is often important to get a good result and to help users
choose the correct model for a data, set the