For genes without annotated transcripts, the RPKM cannot be calculated since the total length of all exons is needed. By checking the Calculate expression for genes without transcripts, the length of the gene will be used in place of an "exon length". If the option is not checked, there will be no RPKM value reported for those genes.
|
Definition of RPKM
RPKM, Reads Per Kilobase of exon model per Million mapped reads, is defined in this way [Mortazavi et al., 2008]:
For prokaryotic genes and other non-exon based regions, the calculation is performed in this way:
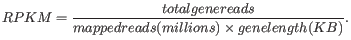
- Total exon reads
- This value can be found in the column with header Total exon reads in the expression track. This is the number of reads that have been mapped to exons (either within an exon or at the exon junction). When the reference genome is annotated with gene and transcript annotations, the mRNA track defines the exons, and the total exon reads are the reads mapped to all transcripts for that gene. When only genes are used, each gene in the gene track is considered an exon. When an un-annotated sequence list is used, each sequence is considered an exon.
- Exon length
- This is the number in the column with the header Exon length in the expression track, divided by 1000. This is calculated as the sum of the lengths of all exons (see definition of exon above). Each exon is included only once in this sum, even if it is present in more annotated transcripts for the gene. Partly overlapping exons will count with their full length, even though they share the same region.
- Mapped reads
- The sum of all mapped reads as listed in the RNA-Seq analysis report. If paired reads were used in the mapping, mapped fragments are counted here instead of reads, unless the Count paired reads as two option was selected. For more information on how expression is calculated in this case, see Calculating expression values from RNA-seq.
|