Find Best References using Read Mapping
To start the tool, go to:
Typing and Epidemiology (![]() ) | Find Best References using Read Mapping (
) | Find Best References using Read Mapping (![]() )
)
The Find Best References using Read Mapping tool takes one or several single or paired-end read sequence lists as input. It then maps simultaneously against a set of references, and optionally a set of host references. The tool will output a list of the references identified. The host references can be used to filter away contaminated reads (e.g. a human reference). The tool can also output the read mappings for both the references and the host.
After specifying the input reads, it is possible to specify the references and adjust the detection parameters, see (figure 11.6).
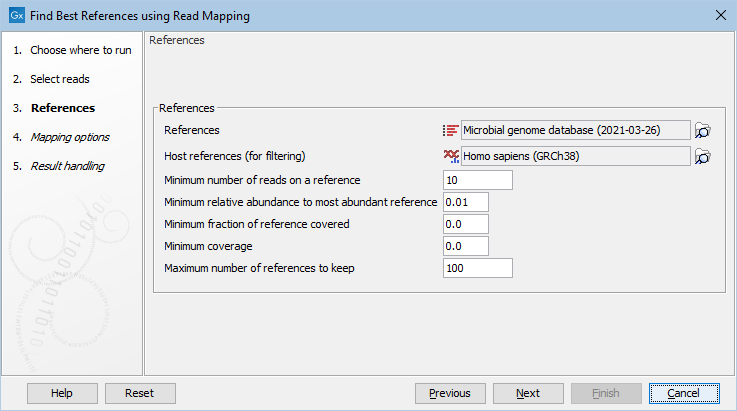
Figure 11.6: Select references and filtering options.
The following parameters are available:
- References Reference sequences as a sequence list or as a sequence track. Notice, that the tool ignores Assembly IDs: each sequence is considered to be its own reference.
- Host references (for filtering) Human reference genome as a sequence list or as a sequence track.
- Minimum number of reads on a reference At least this many reads must map to a reference before it is considered to be present.
- Minimum relative abundance to most abundant reference A reference must have at least this fraction of the reads of the most abundant reference.
- Minimum fraction of reference covered This specifies the minimum fraction of nucleotides in the reference, that must be covered by at least one read.
- Minimum coverage Filters on coverage, which is the number of nucleotides mapped to the reference divided by the reference length.
- Maximum number of references to keep Restricts the number of references to keep. The references are ranked according to the number of hits.
The next step is to specify the read mapping options (figure 11.7). These options are identical to the ones used by the Map Reads to Reference tool.
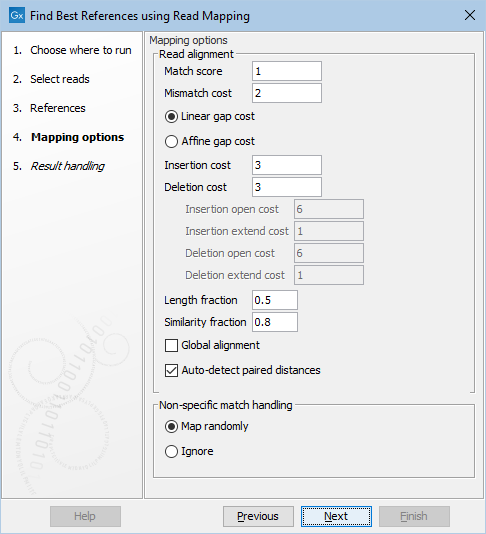
Figure 11.7: Select read mapping options.
The final step is to select which output objects to produce.
Subsections