Create Annotated Sequence List
The Create Annotated Sequence List tool can be used for merging sequences and sequence lists into a single sequence list and/or for annotating the individual sequences with metadata annotations. Metadata annotations are the type of annotations that are visible in columns of the table view of a sequence list, i.e. these annotations are applicable to the whole sequence. This tool can be used to create a variety of databases, e.g. taxonomic and amplicon-based reference databases, gene and resistance databases and many more.
The tool takes any sequence or sequence list as input and outputs a sequence list of nucleotide and/or protein sequences, depending on the input. All nucleotide sequences from the input will be collected into a single output nucleotide sequence list and all protein sequences from the input will be collected into a single protein sequence list.
To run the tool, go to
Tools (![]() ) | Create Annotated Sequence List (
) | Create Annotated Sequence List (![]() ).
).
After selecting which sequences are to be combined and annotated, the annotation sequence sources and general annotation behavior may be specified.
- Overwrite old metadata: If selected, the metadata annotations in existing columns will be overwritten by the specified or downloaded values. Note that missing values (empty strings) will always be replaced with new values if available.
- Annotate with metadata: Transfer metadata from metadata tables in the navigation area that are associated with the input elements onto the individual sequences of the input element, e.g. if a list of contigs is associated with a metadata table that contains the column "Host" with the value "Human", all contigs from this list will be annotated with this column and value in the resulting sequence list. If Overwrite old metadata has been selected, values from the metadata table will be used, otherwise existing values remain on the individual sequences. Note that this kind of metadata may be overwritten by data specified in an external spreadsheet if conflicting columns and values are detected, i.e. the values specified in an external spreadsheet have precedence.
- Download Taxonomy: If selected, the sequences will be decorated with a taxonomy downloaded from NCBI if a TaxID is detected for a sequence. Possible sources for TaxIDs are the "/db_xref="taxon" field in the source annotation of an imported GenBank file and files downloaded with the function "Search for Sequences at NCBI", a "TaxID" column in a metadata table or a "TaxID" column in an external spreadsheet. If a taxonomy already exists, it will only be replaced if Overwrite old metadata has been selected.
- Update latin name: If selected, the latin name of a sequence will be updated from the "/organism" field in the source annotation of an imported .gbff file. If a latin name already exists, it will only be updated if Overwrite old metadata has been selected.
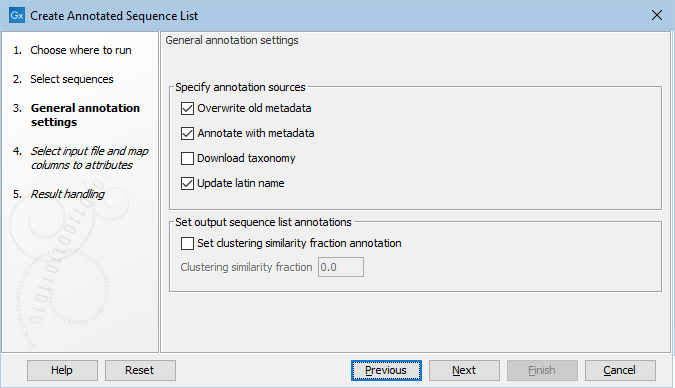
Figure 22.2: The General Annotation Settings of the Create Annotated Sequence List tool.
Sometimes there are empty values in the columns of the incoming data, e.g. in external spreadsheet files or in metadata tables. Whether to overwrite existing values with empty values or rather leave them as they are currently specified can be controlled in the Overwrite with empty values section by selecting Update with empty values or Ignore empty values, respectively.
Optionally annotations may be set for the whole sequence list, e.g. if the sequence list contains sequences clustered at a specific sequence similarity, this may be set under Clustering similarity fraction after checking Set Clustering similarity fraction annotation. These options are for example required when constructing an amplicon-based reference database.
The next wizard step makes it possible to optionally specify an Excel spreadsheet or plain text file with annotations for the sequences. In the Import section an Excel or plain text file can be specified. As soon as a file has been selected it will appear in the preview table in the bottom of the wizard. Excel files are straight-forwardly recognized, while it may be necessary to set the Encoding, Separator and Quote symbols for plain text files in order to parse them, the preview will be updated according to how the file is read with the specified settings. Note that the tool matches names found in a column called "Name" with the names of the sequences to transfer the specified values, which means that a column called "Name" has to be specified.
- Encoding: Specify the encoding for the selected file. This option is not available for Excel files.
- Offset: Control how many lines will be skipped at the beginning of the file.
- Separator: Specify the character used to separate columns in the selected file. This option is not available for Excel files.
- Quote symbol: Specify the character used to define a string. This is typically used if a field value may contain the character used to separate the columns. This option is not available for Excel files.
- Handle duplicates: This parameter controls how entries with the same name in the input file are treated. Available options are to use "Only last entry", to "Concatenate entries" or to "Concatenate unique entries". Note that the way the entries are concatenated may depend on the specified metadata field. The standard concatenation is done using a comma followed by a space between entries.
- Column to match: This parameter controls which column shall be used for name matching. All rows for which the entries match will be updated with the new values.
- Named columns: Controls whether the first line is a header line or part of the data.
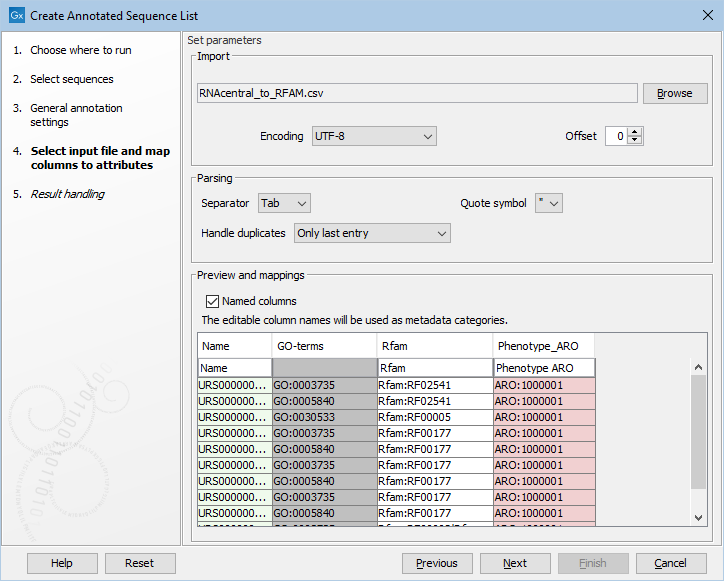
Figure 22.3: Options for annotating a sequence list with an external file.
The preview table in the bottom will be limited to 100 lines or 1MB of the input file and the first line can be modified to specify how the columns should be mapped to metadata annotations. A column named "Name" is required to be able to match a table row with a sequence, other columns may have a special meaning, indicated by the color of the column in the preview. Possible colors are
- Green: The specified column header has a special meaning, where the name matching to metadata annotation fields is case-insensitive.
- Red: The specified column header has a special meaning, but at least one of the values in the displayed preview for this column does not match the expected format of the metadata annotation field, or this kind of metadata cannot be changed (e.g. 'Size' or 'Start of sequence'). These values will be skipped.
- Gray: No header has been specified. This column will be ignored.
- White: No special meaning associated with the specified header.
The most important header names with special meaning are
- Name: The only required column, specifying the name of the sequence the metadata annotations will be transferred to.
- Taxonomy: Specifies the taxonomy for a given sequence. This field is for example used by Create Taxonomic Profiling Index and downstream tools and visualizations.
- TaxID: This field specifies the TaxID for a given sequence and may be used to populate the Taxonomy field if "Download Taxonomy" has been selected.
- Assembly ID: This field is used to group sequences into logical units. For more information on how this field is used, please see section Assembly ID.
Subsections