Example of results obtained using the Type Among Multiple Species workflow
The following example includes typing of 2 samples: 1 Salmonella enterica (acc no. ERR277212), and 1 Yersinia ruckeri (acc no SRR3152422). Using the workflow Type Among Multiple Species workflow, analysis results are automatically summarized in the Result Metadata Table as shown in figure 24.43. The analysis results in this example include resistance found for antibiotic inactivation enzyme, name of the best matching reference, applied MLST scheme, detected sequence type and typing status. You could also choose (using options in the Table Setting window) to display additional information such as the 'Best Match, Species' and 'Find Resistance With Nucleotide DB' for example.
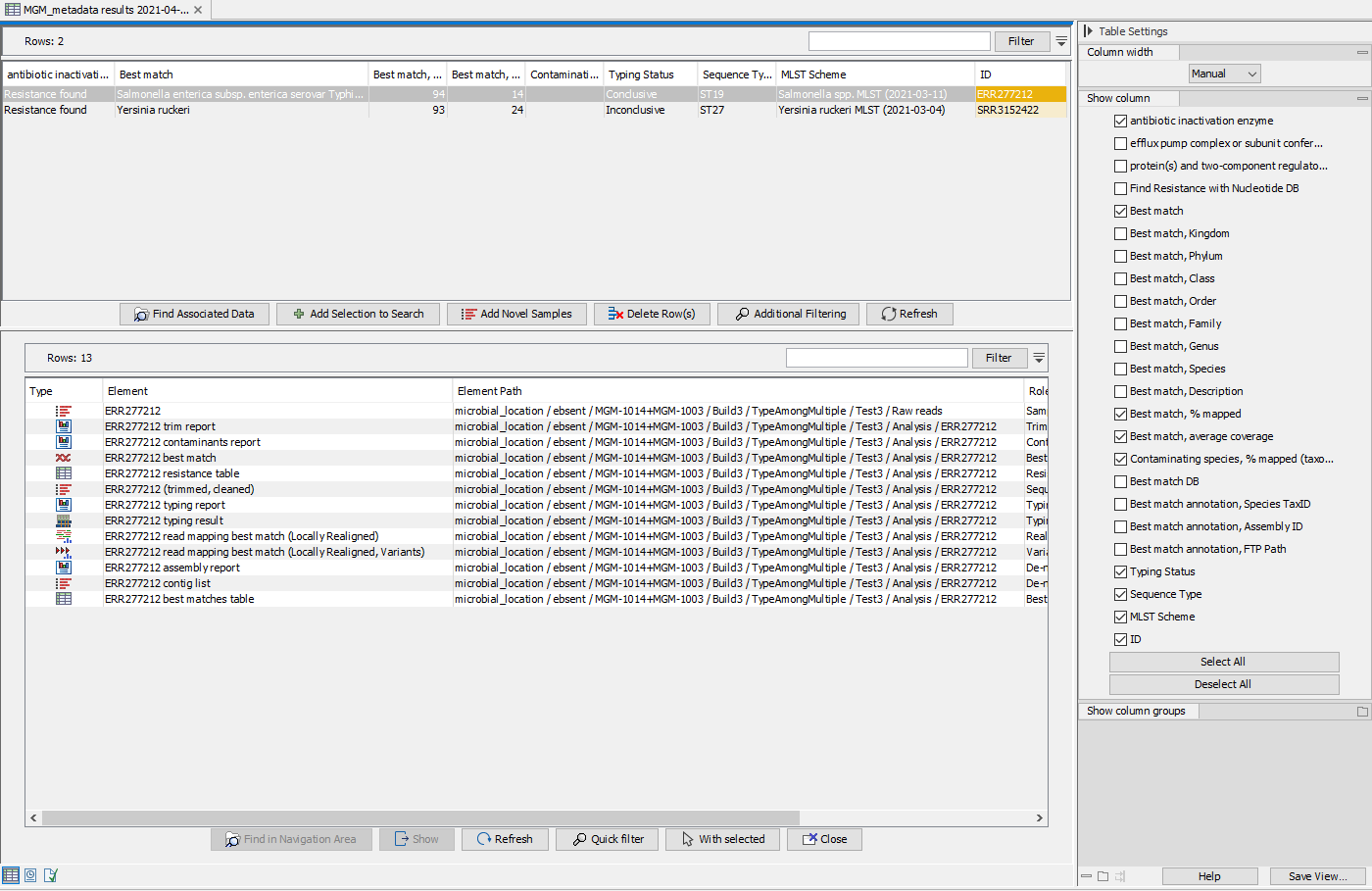
Figure 10.22: View of Result Metadata Table once the Type Among Multiple Species workflow has been executed (top) and associated data elements found (bottom).
Analyzing samples in batch will produce a large amount of output files, making it necessary to filter for the information you are looking for. Through the Result Metadata Table, it is possible to filter among sample metadata and analysis results. By clicking Find Associated Data (![]() ) and optionally performing additional filtering, it is possible to perform additional analyses on a selected subset directly from this Table, such as:
) and optionally performing additional filtering, it is possible to perform additional analyses on a selected subset directly from this Table, such as:
- Generation of SNP trees based on the same reference used for read mapping and variant detection (Create SNP Tree).
- Generation of K-mer Trees for identification of the closest common reference across samples (Create K-mer Tree).
- Run validated workflows (workflows that are associated with a Result Metadata Table and saved in your Navigation Area).
Note that the tool will output, among other files, variant tracks. It is possible to export multiple variant track files from monoploid data into a single VCF file with the Multi-VCF exporter.This exporter becomes available when installing the CLC Microbial Genomics Module. All variant track files must have the same reference genome for the Multi-VCF export to work.