Compare Variants Across Samples
The Compare Variants Across Samples can be used to compare samples originating from strains or species sharing a common reference. The workflow takes trimmed, host-filtered reads as input such as output produced by the Data QC and Remove Background Reads, Type a Known Species, Type Among Multiple Species or Analyze Viral Hybrid Capture Panel Data workflow. As the workflow removes duplicate mapped reads, amplicon data is not recommended as input. However, the workflow can be modified to work on amplicon data by opening a copy of the workflow, removing the Remove Duplicate Mapped Reads tool and saving the modified workflow.
To run the Compare Variants Across Samples workflow, go to
Microbial Genomics Module (![]() ) | Typing and Epidemiology (
) | Typing and Epidemiology (![]() ) | Workflows (
) | Workflows (![]() ) | Compare Variants Across Samples (
) | Compare Variants Across Samples (![]() ).
).
An overview of the workflow can be seen in figure 10.42.
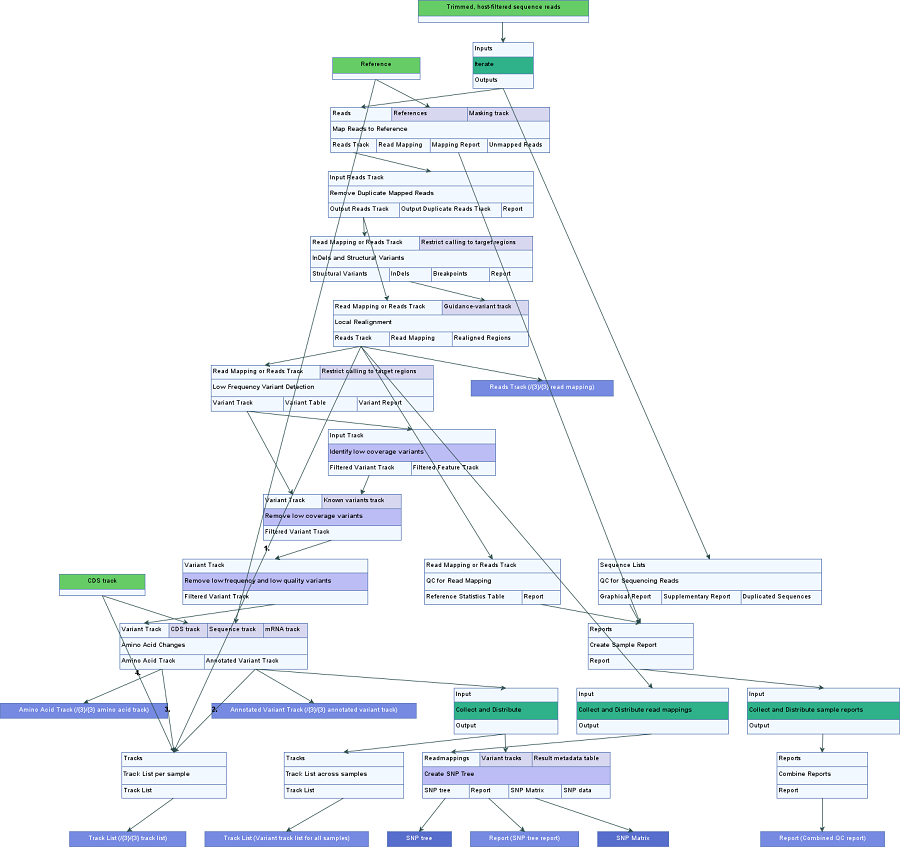
Figure 10.42: An overview of the Compare Variants Across Samples workflow
- Select two or more read sets as input (figure 10.43). The workflow uses internal batching and creates an analysis for each sample as well as a combined variant track and SNP tree.
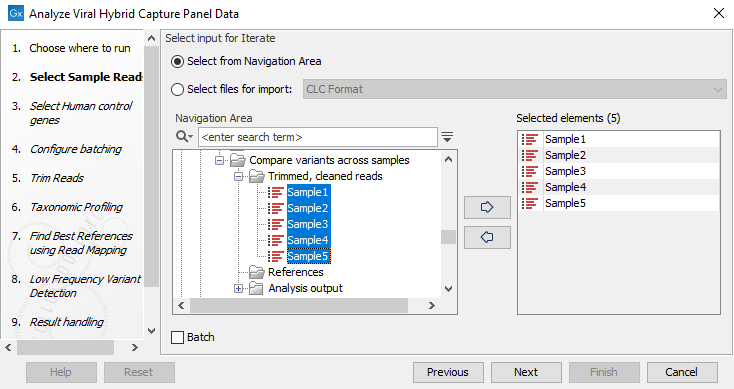
Figure 10.43: Select input data with common reference for analysis - Select the reference to use (figure 10.44). The reference should match all the samples selected.
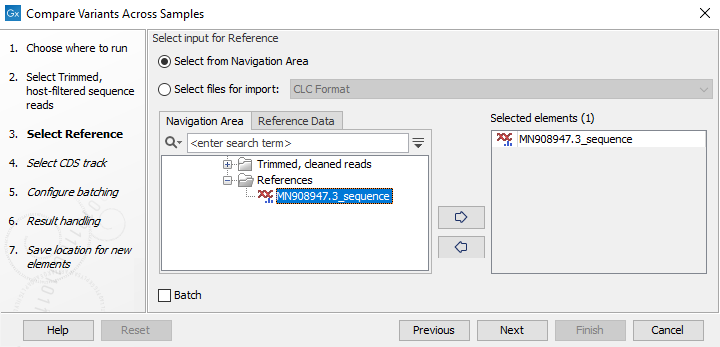
Figure 10.44: Select reference - Select a CDS track associated with the reference (figure 10.45).
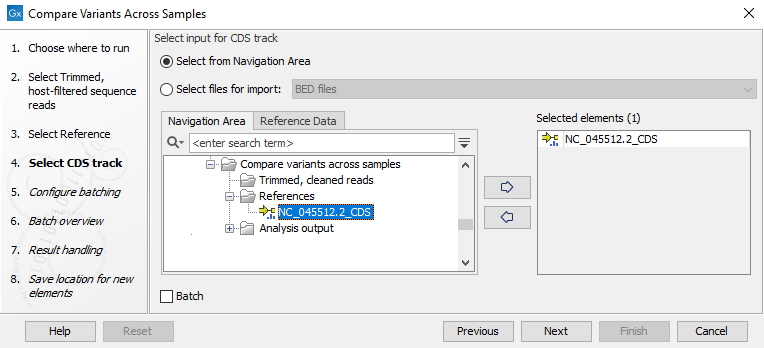
Figure 10.45: Select CDS track used to annotate variants - In the Result handling window, pressing the button Preview All Parameters allows you to preview - but not change - all parameters.
Saving the workflow output will generate the files shown in (figure 10.46) and optionally, a workflow result metadata table.
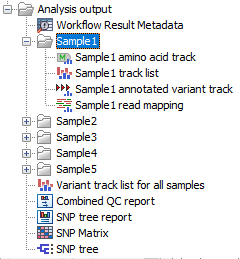
Figure 10.46: Output from Compare Variants Across Samples workflow
The workflow generates outputs for each batch analysis run as well as a folder for each sample. For each sample, the following is output:
- Annotated variant track: output from the Low Frequency Variant Detection tool after coverage and quality filtering. Note that it is possible to export multiple variant track files from monoploid data into a single VCF file with the Multi-VCF exporter. This exporter becomes available when installing the CLC Microbial Genomics Module. All variant track files must have the same reference genome for the Multi-VCF export to work.
- Amino acid track: amino acid track including amino acid changes resulting from the called variants.
- Read mapping: output from the Local Realignment tool, mapping of the reads to the specified reference. For increased sensitivity, duplicate mapped reads are removed before local realignment.
- Track list: output from the Create Track List tool. The track list combines the read mapping, variant, amino acid and CDS tracks. An example can be seen in figure 10.47.
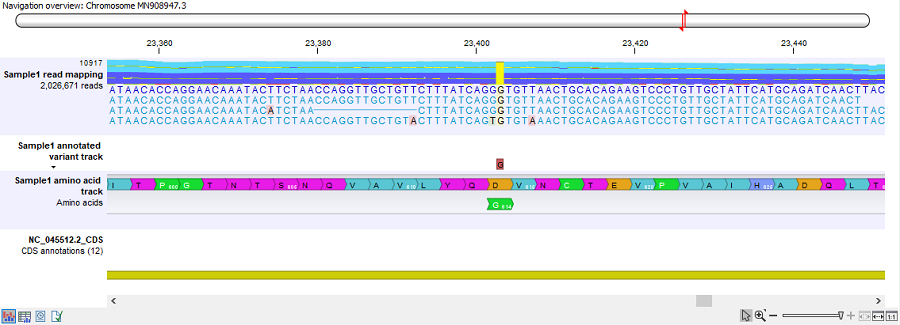
Figure 10.47: The track list generated for each sample analysis
For each batch analysis run, the following outputs are generated:
- Variant track list for all samples: output from the Create Track List tool. The track combines the variant tracks for all analyzed samples.
- Combined QC report: a combined report built from QC for sequencing reads, Read mapping summary and QC for read mapping. This report contains a summary of all analyzed samples.
- SNP tree report: summarizes the consequence of the applied filtering settings in the Create SNP tree tool, as well as a summary of ignored positions attributed to the different read mappings.
- SNP matrix: a matrix containing the pairwise number of SNP differences between all pairs of samples included in the analysis (see figure 10.48).
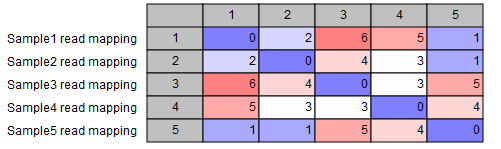
Figure 10.48: SNP matrix for pairwise comparisons of all samples included in the analysis - SNP tree: the output tree built from the SNPs called in all samples (see figure 10.49). A number of different visualizations are available, see Visualization of SNP Tree including metadata and analysis result metadata.
Here, the leaf nodes have been colored according to geographic location of the collected samples.
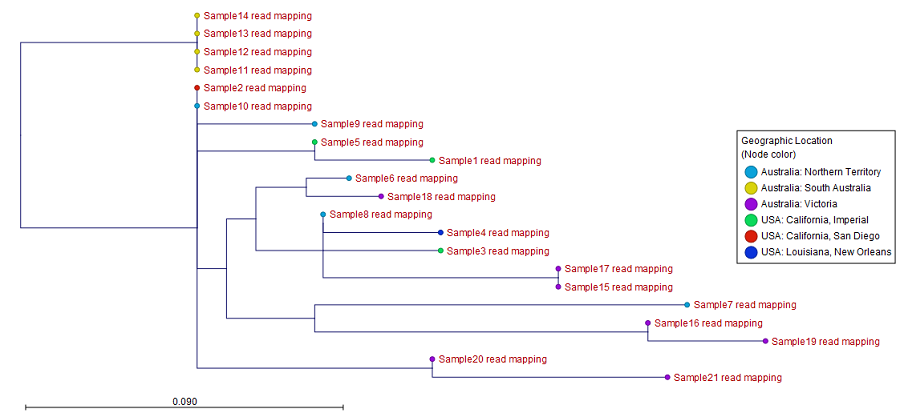
Figure 10.49: An output SNP tree of 21 samples with leaf nodes colored using metadata annotation
For more information on the tree tools, see Create SNP Tree.