Remove duplicate mapped reads
The purpose of this tool is to efficiently remove duplicate reads from a mapping, when duplicate reads have arisen due to the use of PCR amplification (or other enrichment) during sample preparation. This does not mean, however, that this tool should be used on all data that had an amplification step. In fact, use of this tool in the case of RNA-Seq data, amplicon data, or any sample where the start of a large number of reads are purposely at the same reference location, it is not recommended to make use of this tool. The tool may be used on mappings of single end reads, paired end reads or both.
A read duplication can be easily distinguished when mapping reads to a reference sequence as shown in the example in figure 19.28.
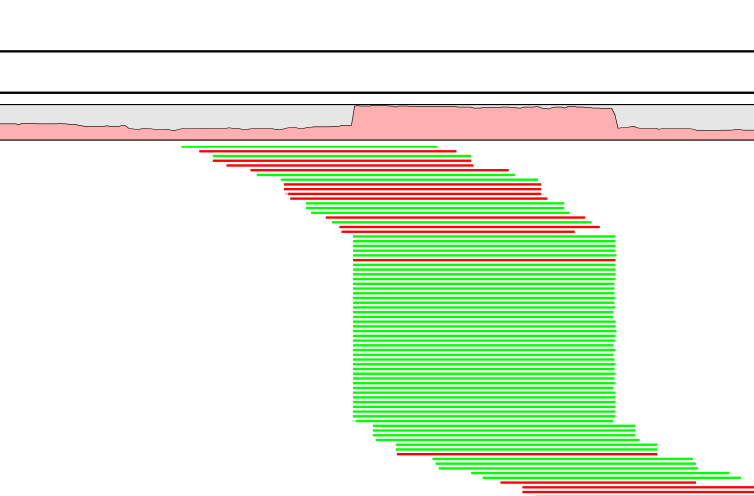
Figure 19.28: Mapped reads with a set of duplicate reads, the colors denote the strand (green is forward and red is reverse).
When sequencing library preparation involves a PCR amplification step, it is common to observe multiple reads where identical nucleotide sequences are disproportionably represented in the final results. Thus, to facilitate processing of mappings based on this kind of data, it may be necessary to perform a duplicate read removal step, which flags identical reads and subsequently removes them from the data set. However, this step is complicated by the low, but consistent, presence of sequencing errors that may cause otherwise identical sequences to differ slightly. Thus, it is important that the duplicate read removal includes some tolerance for nearly identical sequences, which could still be reads from the same PCR artifact.
In samples that have been mapped to a reference genome, duplicate reads from PCR amplification typically result in areas of disproportionally high coverage and are often the cause of significant skew in allelic ratios, particularly when replication errors are made by the enzymes (e.g. polymerases) used during amplification. Sequencing errors incorporated post-amplification can affect both sequence- and coverage-based analysis methods, such as variant calling, where introduced errors can create false positive SNPs, and ChIP-Seq, where artificially inflated coverage can skew the significance of certain locations. By utilizing the mapping information, it is possible to perform the duplicate removal process rapidly and efficiently.
Note! We only recommend using the duplicate read removal if there are amplification steps involved in the library preparation. It is not reccomended for RNA-Seq data, amplicon data, or any sample where the start of a large number of reads are purposely at the same reference location.
The method used by the duplicate read removal is to identify reads that share common coordinates (e.g. the same start and end coordinate), sequencing direction (or mapped strand) and the same sequence, these being the unifying characteristics behind sequencing reads that originate from the same amplified fragments of nuclear material. However, due to the frequent occurrence of sequencing errors, the tool utilizes simple heuristics to prune sequences with small variations from the consensus, as would be expected from errors observed in data from next-generation sequencing platforms. Base mismatch errors that were incorporated during amplification or prior to amplification will be indistinguishable from SNPs and may not be filtered out by this tool.
Subsections