Identify variants and add expression values
The Identify Variants and Add Expression Values ready-to-use workflows can be used to identify novel and known mutations in RNA-seq data, automatically map, quantify, and annotate the transcriptomes, and compare the mutational patterns in the samples with the expression values of the corresponding transcripts and genes.
To run the ready-to-use workflow:
Toolbox | Ready-to-Use Workflows | Whole Transcriptome Sequencing (![]() ) | (Human (
) | (Human (![]() ), Mouse (
), Mouse (![]() ) or Rat (
) or Rat (![]() )) | Identify Variants and Add Expression Values (
)) | Identify Variants and Add Expression Values (![]() )
)
- Double-click on the Identify Variants and Add Expression Values tool to start the analysis. If you are connected to a server, you will first be asked, where you would like to run the analysis.
- Specify the RNA-seq reads to analyze. The reads can be selected by double-clicking on the reads file name or clicking once on the file and then clicking on the arrow pointing to the right side in the middle of the wizard (figure 15.25).
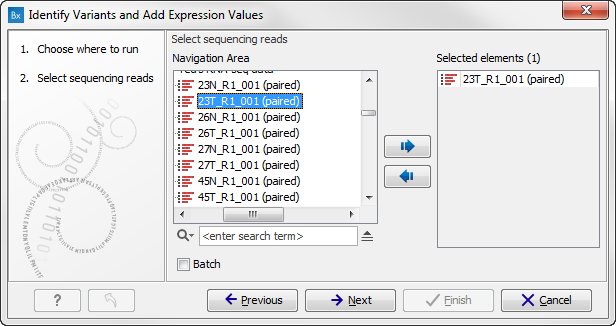
Figure 15.25: Select the sequencing reads to analyze.Click on the button labeled Next.
- Specify a target region for the Indels and Structural Variants tool (figure 15.26).
The targeted region file is a file that specifies which regions have been sequenced. This file is something that you must provide yourself, as this file depends on the technology used for sequencing. You can obtain the targeted regions file from the vendor of your targeted sequencing reagents. Remember that you have a hg38-specific BED file when using hg38 as reference, and hg19-specific BED file when using hg19 as reference.
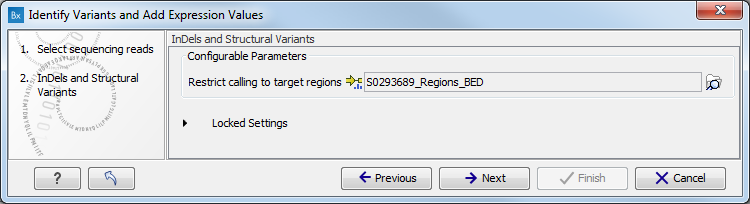
Figure 15.26: Specify the target region for the Indels and Structural Variants tool. - Set the parameters for the Low Frequency Variant Detection step (see figure 15.27). For a description of the different parameters that can be adjusted in the variant detection step, see http://clcsupport.com/biomedicalgenomicsworkbench/current/index.php?manual=Low_Frequency_Variant_Detection.html.
If you click on "Locked Settings", you will be able to see all parameters used for variant detection in the ready-to-use workflow.
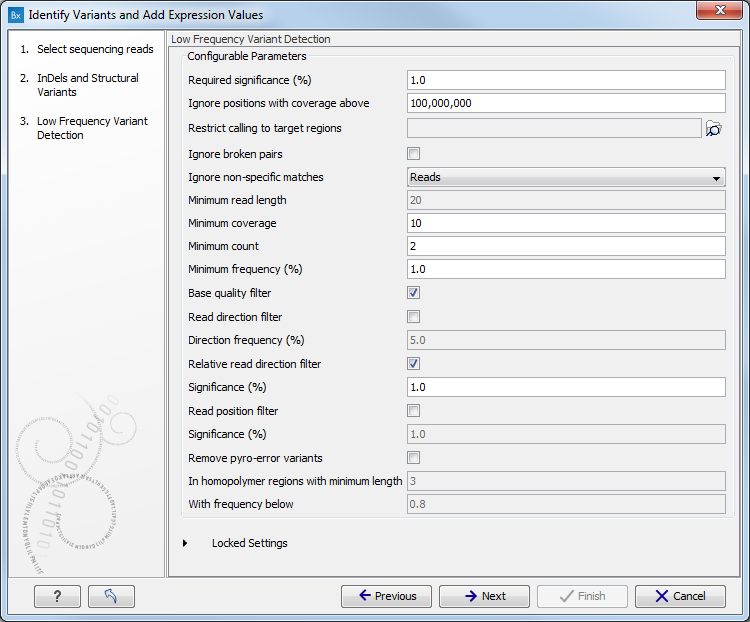
Figure 15.27: Specify the parametes for transcriptomic variant detection. - If you are working with the workflow from the Human folder, specify here the relevant 1000 Genomes population from the drop-down list (see figure 15.28). Choose the population that matches best the population your samples are derived from.
Under "Locked settings" you can see that "Automatically join adjacent MNVs and SNVs" has been selected. The reason for this is that many databases do not report a succession of SNVs as one MNV as is the case for the Biomedical Genomics Workbench, and as a consequence it is not possible to directly compare variants called with Biomedical Genomics Workbench with these databases. In order to support filtering against these databases anyway, the option to Automatically join adjacent MNVs and SNVs is enabled. This means that an MNV in the experimental data will get an exact match, if a set of SNVs and MNVs in the database can be combined to provide the same allele.
Note! This assumes that SNVs and MNVs in the track of known variants represent the same allele, although there is no evidence for this in the track of known variants.
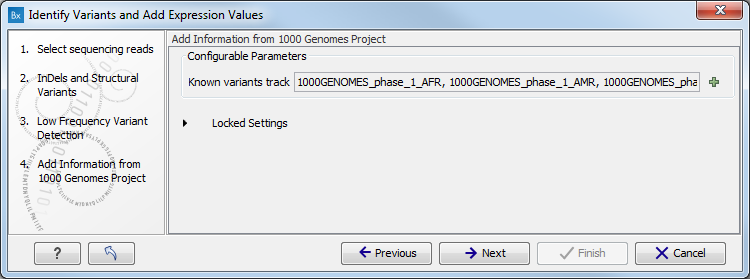
Figure 15.28: Select the relevant population from the drop-down list. - Click on the button labeled Next to go to the last wizard step (shown in figure 15.29).
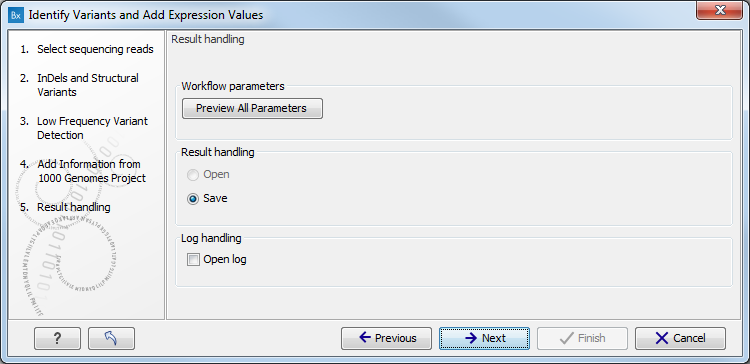
Figure 15.29: Check the selected parametes by pressing "Preview All Parameters".Pressing the button Preview All Parameters allows you to preview all parameters. At this step you can only view the parameters, it is not possible to make any changes (see figure 15.30). Choose to save the results and click on the button labeled Finish.
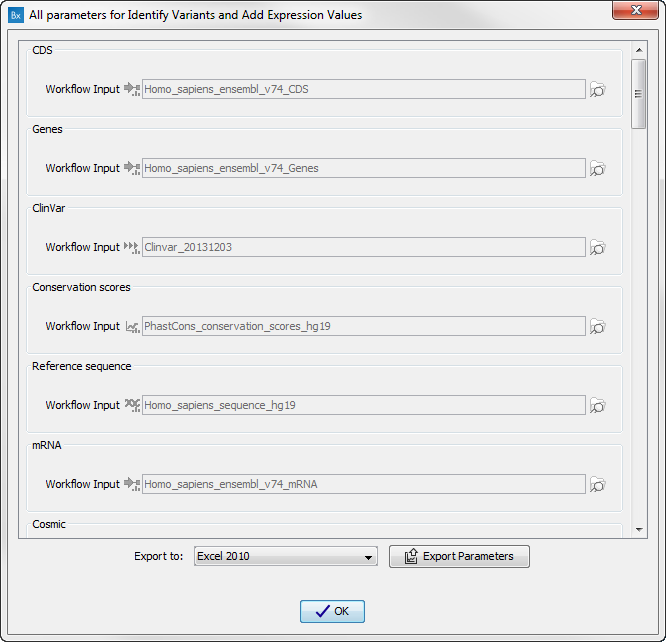
Figure 15.30: Preview all parameters. At this step it is not possible to introduce any changes, it is only possible to view the settings.
Seven different output types are generated:
- Gene expression (
 ) A track showing gene expression annotations. Hold the mouse over or right-clicking on the track. A tooltip will appear with information about e.g. gene name and expression values.
) A track showing gene expression annotations. Hold the mouse over or right-clicking on the track. A tooltip will appear with information about e.g. gene name and expression values.
- Transcript expression () A track showing transcript expression annotations. Hold the mouse over or right-clicking on the track. A tooltip will appear with information about e.g. gene name and expression values.
- RNA-Seq Mapping Report (
 ) This report contains information about the reads, reference, transcripts, and statistics. This is explained in more detail in the Biomedical Genomics Workbench reference manual in section RNA-Seq report (http://clcsupport.com/biomedicalgenomicsworkbench/current/index.php?manual=RNA_Seq_report.html).
) This report contains information about the reads, reference, transcripts, and statistics. This is explained in more detail in the Biomedical Genomics Workbench reference manual in section RNA-Seq report (http://clcsupport.com/biomedicalgenomicsworkbench/current/index.php?manual=RNA_Seq_report.html).
- Read Mapping (
 ) The mapped RNA-seq reads. The RNA-seq reads are shown in different colors depending on their orientation, whether they are single reads or paired reads, and whether they map unambiguously. For the color codes please see the description
in (see http://www.clcsupport.com/biomedicalgenomicsworkbench/current/index.php?manual=View_settings_in_Side_Panel.html).
) The mapped RNA-seq reads. The RNA-seq reads are shown in different colors depending on their orientation, whether they are single reads or paired reads, and whether they map unambiguously. For the color codes please see the description
in (see http://www.clcsupport.com/biomedicalgenomicsworkbench/current/index.php?manual=View_settings_in_Side_Panel.html).
- Annotated Variants with Expression Values (
 ) Annotation track showing the variants. Hold the mouse over one of the variants or right-clicking on the variant. A tooltip will appear with detailed information about the variant.
) Annotation track showing the variants. Hold the mouse over one of the variants or right-clicking on the variant. A tooltip will appear with detailed information about the variant.
- RNA-Seq Genome Browser View (
 ) A collection of tracks presented together. Shows the annotated variants track together with the human reference sequence, genes, transcripts, coding regions, and variants detected in ClinVar and dbSNP (see figure 15.15).
) A collection of tracks presented together. Shows the annotated variants track together with the human reference sequence, genes, transcripts, coding regions, and variants detected in ClinVar and dbSNP (see figure 15.15).
- Log (
 ) A log of the workflow execution.
) A log of the workflow execution.