The concept of CLC Single Cell Analysis Module
CLC Single Cell Analysis Module 21.1 enables the study of single cell RNA (scRNA-seq) and T-cell receptor (scTCR-seq) samples. The comprehensive toolbox includes tools for analyzing the two types of data both separately and jointly. The Single Cell Analysis toolbox is shown in figure 1.1
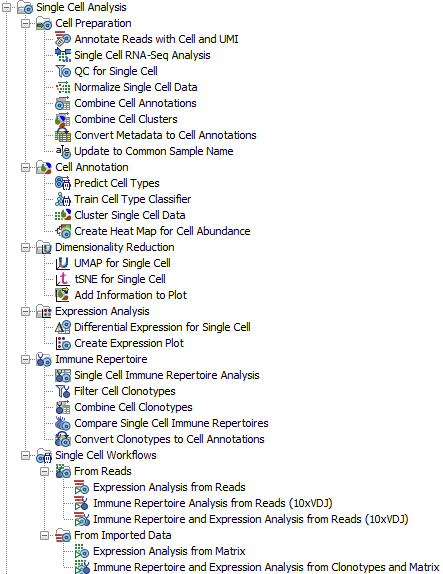
Figure 1.1: The Single Cell Analysis toolbox with tools for analyzing scRNA-seq and scTCR-seq data from sample to insight.
scRNA-seq
Tools are available for quality control and normalization, noise reduction and feature selection, clustering, cell type prediction, and differential expression. UMAP and tSNE plots can be overlaid with clusters, predicted cell types, or the expression of individual genes. Marker genes can be identified through analyses of differential gene expression and by gathering information from expression plots. Alternatively, cell type classifiers can be trained from pre-labeled cells. Two pre-trained classifiers are provided, and can be extended. The provided workflows can be easily adjusted to fit the chemistry and protocol of the data and are a good starting point from either raw FASTQ or an imported Expression Matrix.
scTCR-seq
After the initial identification of clonotypes in the sample, these can be further filtered, combined across samples and the sample-level immune repertoires can be compared with regards to diversity estimates, gene usage, etc. UMAP and tSNE plots from matched scRNA-seq data can be overlaid with clonotype information, once this is converted to Cell Annotations. The provided workflows are a good starting point from either raw FASTQ or imported Cell Clonotypes. Additionally, workflows are available for the joint analysis of both scRNA-seq and scTCR-seq data.
Selection of algorithms
The algorithms implemented have been selected to be the best performing at the time of development as assessed by independent paper reviews. All algorithms have been re-implemented in Java with the aim of being able to scale to large data sets and run on a wide range of hardware. Internal benchmarks have been conducted to select the best performing algorithm for predicting cell types, which is one of the key features in this software package. The manual provides detailed descriptions of the chosen algorithms, how to adjust parameters for better performance, and how to interpret results.
Manual content
The manual contains multiple parts, that each address a different aspect of the software package. A short description of each part follows:- Introduction Contains basic information needed for installing the module and handling licenses. It describes reference data and how to run tools and workflows. This part is less relevant for experienced users.
- Import and Export Contains information on the wide range of supported expression matrix formats and how to import and export data to and from third party tools.
- Single Cell Analysis This section is divided into chapters covering creating a count matrix, cell preparation, noise reduction, cell annotation, dimensionality reduction, expression analysis, and immune repertoire analysis. Each of these chapters describes the algorithms behind the tools or approaches as well as how to select parameters. These sections are valuable if unexpected results are obtained from using default parameters, and provide detailed insight into the algorithms.
- Prebuilt workflows Provides descriptions of the workflows and how adjustments should be made to match the chemistry and sequencing protocol of the samples in question.
- Tips and tricks In this part you get all the hidden gems. Here you can learn about the functionality built into the different plots and visualizations, and how you can manipulate them to better fit your needs. You can also learn about the QIAGEN Gene Ontology browser that can be used to access additional information about your cell types. We recommend that advanced users start the journey here.