The Normalize Single Cell Data algorithm
The algorithm is based on sctransform [Hafemeister and Satija, 2019]. Briefly, a negative binomial (NB) generalized linear model (GLM) is fit to
where ![]() are the observed counts for the gene for a cell
are the observed counts for the gene for a cell ![]() that has
that has ![]() total counts. The dispersion parameter
total counts. The dispersion parameter
![]() of the NB distribution is estimated during fitting using the Cox-Reid penalized adjusted likelihood [Robinson et al., 2010]. When
of the NB distribution is estimated during fitting using the Cox-Reid penalized adjusted likelihood [Robinson et al., 2010]. When ![]() (
(
![]() ) the NB distribution reduces to the Poisson distribution.
) the NB distribution reduces to the Poisson distribution.
LOWESS regression is then used to estimate the intercept ![]() , the log-sequencing-depth coefficient
, the log-sequencing-depth coefficient ![]() , and the dispersion as a function of the average expression. The regression serves as a form of regularization that avoids over-fitting the model, which happens especially for low expression genes.
, and the dispersion as a function of the average expression. The regression serves as a form of regularization that avoids over-fitting the model, which happens especially for low expression genes.
For batch correction there are some differences from both the above and from sctransform:
- A GLM is fit to every gene, because any gene might have a batch effect - though genes with expression
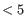 counts are ignored.
counts are ignored.
- Batch effect terms are added to the model, and these cannot be regularized because each gene might have a batch effect very different from those of genes with similar expression.
- LOWESS regression is only applied to the dispersion, because otherwise there is a mix of regularized and non-regularized terms that cannot be disentangled. The end result is, roughly speaking, that the price to pay for batch correction is a tendency to over-correct data. However, the more data there is, the less this will be a problem, and batch correction is typically performed on large amounts of data.
Normalized/batch corrected values are Pearson Residuals. For each gene, these are defined as follows:
Note that Pearson Residuals have several properties that may be unexpected. They are:
- decimals e.g. 123.4 rather than integers e.g. 123
- negative when a gene in a cell has lower expression than predicted by the underlying GLM (though usually not very negative)
- zero for all cells in the unlikely event that expression can be perfectly predicted by the GLM from the provided combination of sequencing depth and batch factors
- only defined in the context of a data set - they cannot be compared across data sets. For example if we have three data sets A, B and C, then running the tool on (A + B) might say that a particular cell + gene in set A has a normalized expression of 100, while running the same tool on (A + C) might say that the same cell + gene has a normalized expression of 0.