Normalize Single Cell Data
The Normalize Single Cell Data tool transforms count data so as to remove the effect of sequencing depth and, optionally, the effect of batch factors. It is recommended to use this tool prior to downstream analysis.
Normalize Single Cell Data can be found in the Toolbox here:
Cell Preparation (![]() ) | Normalize Single Cell Data (
) | Normalize Single Cell Data (![]() )
)
The tool takes at least one Expression Matrix (![]() ) as input, and produces a single Expression Matrix as output. If multiple Expression Matrixes are provided as input, the single Expression Matrix output will be filtered to only contain those genes that are present in all of the inputs. A report can optionally also be output.
) as input, and produces a single Expression Matrix as output. If multiple Expression Matrixes are provided as input, the single Expression Matrix output will be filtered to only contain those genes that are present in all of the inputs. A report can optionally also be output.
There are three ways of using the Normalize Single Cell Data tool, which differ in how batch correction is performed:
- None. Batch correction is not applied, but count data is transformed so as to remove the effect of sequencing depth. For a new dataset, it is often sensible to first try this setting, and then only apply a batch correction if a batch effect is evident in the Dimensionality Reduction Plot. For more details see When is batch correction appropriate?.
- Each sample is a batch. Batch correction is performed by choosing one sample as the `baseline'. Transformations for each additional sample are applied to make them resemble the baseline. This is appropriate when each sample is expected to have systematic changes in gene expression compared with all other samples, and when these changes are uninteresting for downstream analysis. For example, this setting may be appropriate for combining samples of the same tissue created by different investigators.
- Using metadata. A flexible batch correction is applied, where each batch can consist of several inputs (Sample level metadata), or where batches can be specified at the level of individual cells (Cell level metadata).
Sample level metadata can be supplied as a Metadata table. For details on how to create a Metadata Table, see https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Metadata.html.To use sample level metadata, multiple inputs must be provided, because each batch will consist of at least one input.
Batch factors can be supplied in the Correct for field. These correspond to columns of a Metadata table. The use of more than one batch factor is not advised as it is easy to over-parameterize the model, see Interpreting the output of Normalize Single Cell Data.
It is also possible to supply Do not correct for factors. When these are present, the tool will warn if correcting for the specified batch effect would remove all variation due to these factors in at least one sample (because they are confounded). It will also explicitly model the effect of these factors on expression, which helps to prevent variation due to these factors from being removed by the batch effect correction. This should be regarded as `advanced' functionality because it is easy to over-parameterize the model, see Interpreting the output of Normalize Single Cell Data.
A typical use case for sample level metadata might be when combining samples of the same tissue prepared by different investigators, but where each investigator might have prepared multiple samples. Here it would make sense to `Correct for = investigator'. If each investigator prepared a mixture of treated and control samples, then it would make sense to `Correct for = investigator' and `Do not correct for = Treatment/Control'.
Cell level metadata Batch factors can also be specified from columns in Cell Clusters and Cell Annotations. Numerical columns of Cell Annotations are not supported, so it is not possible to, for example, regress out `Mitochondrial counts (%)', but this practice is also not advised [Germain et al., 2020]. Multiple inputs of each type are supported, so it is not necessary to `combine' Cell Clusters and Cell Annotations before the tool is run.
It is easiest to explain the batch correction process with an example. If correcting for a cell cycle annotation, possible values might be "G2/M, G1, S". Cells without an annotated cell cycle state, or with an annotation that is rare (shared by
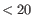 cells) are given an additional value "Unknown". One of these four cell cycle states (G2/M, G1, S1, Unknown) is then chosen as the baseline, and transformations for each additional value are applied to make the other cell cycle states resemble the baseline.
cells) are given an additional value "Unknown". One of these four cell cycle states (G2/M, G1, S1, Unknown) is then chosen as the baseline, and transformations for each additional value are applied to make the other cell cycle states resemble the baseline.
Subsections
- When is batch correction appropriate?
- Interpreting the output of Normalize Single Cell Data
- The Normalize Single Cell Data algorithm