General notes on handling paired data
During import, information about paired data (distances and orientation) can be specified (see figure 7.10) and stored by the CLC Genomics Workbench. All subsequent analyses automatically take differences in orientation into account. Once imported, both reads of a pair will be stored in the same sequence list. The forward and reverse reads (e.g. for paired-end data) simply alternate so that the first read is forward, the second read is the mate revere read; the third is again forward and the fourth read is the mate reverse read and so on. When manipulating sequence lists with paired data, be careful not break this order.
You can view and edit the orientation of the reads after they have been imported by opening the read list in the Element information view (![]() ), as shown in figure 7.18.
), as shown in figure 7.18.
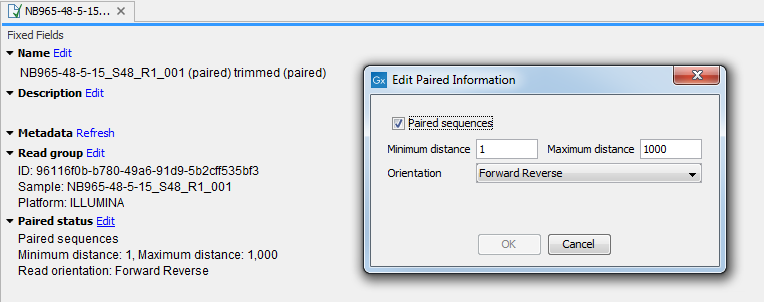
Figure 7.18: The paired orientation and distance.
In the Paired status part, you can specify whether the CLC Genomics Workbench should treat the data as paired data, what the orientation is and what the preferred distance is. The orientation and preferred distance is specified during import and can be changed in this view. If the "Paired sequences" box is unchecked, the sequences will be handled as single (non paired) data.
Note that the paired distance measure that is used throughout the CLC Genomics Workbench is always including the full read sequence. For paired-end libraries it means from the beginning of the forward read to the beginning of the reverse read.