Sample reads
The Sample Reads tool is very useful for reducing the size of data sets. It allows the user to reduce the number of reads in a sequence list to an absolute number of reads, or a pre-defined percentage of the original list.
A reduced sequence list can be relevant when editing contigs with a high coverage, as an excessive amount of reads can be overly resource-demanding and can hinder read mapping editor functions. Sampling reads, and subsequently mapping the sampled reads to contigs with the Map Reads to Contig tool can maintain the speed of many edit operations in the contig aligner without any loss of important information. Similarly, in the case of de novo assembly, very high coverage in a location will increase the probability this location is seen as a sequencing error. You can thus use the Sample Reads tool to reduce coverage to a maximum average coverage of 100x using the following calculation:
- Multiply the estimated size of the genome you intend to assemble by 100. This value will be the total number of bases you should use as input for assembly.
- Divide the total input bases the average length of your sequencing reads.
- Use this number as input for the number of reads to obtain as output from the Sample Reads tool.
To run the Sample Reads tool:
Toolbox | Utility Tools (![]() ) | Sample reads (
) | Sample reads (![]() )
)
This opens the dialog shown in figure 34.11. Select the relevant reads and click Next.
Figure 34.11: Select the reads.
This leads to the Sample size parameters step shown in figure 34.12.
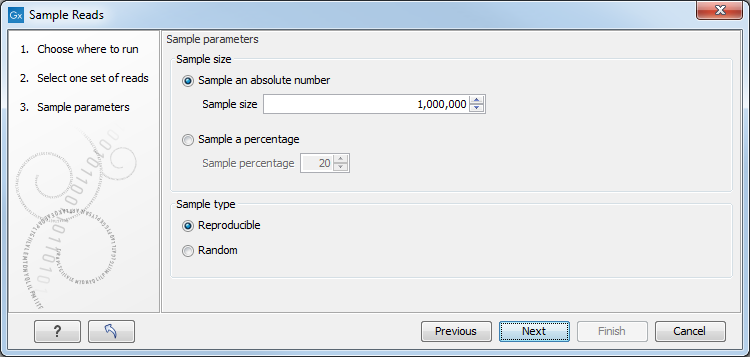
Figure 34.12: Specify the sample size parameters.
In the Sample size parameters it is possible to sample down to either an absolute number of reads, or a sample percentage of the original file, with sample percentage defining the percentage of the sub-sample paired reads (for single end reads) and pairs of reads (for paired reads).
It is also possible to choose between a Reproducible or Random output. The Reproducible option will always returns the same sample (i.e., containing the same reads) for a given sample size or percentage; the Random option returns a different sample each time the tool is run.
After the Result handling step, click Finish.