BLAST at NCBI
When running a BLAST search at the NCBI, the Workbench sends the sequences you select to the NCBI's BLAST servers. When the results are ready, they will be automatically downloaded and displayed in the Workbench. When you enter a large number of sequences for searching with BLAST, the Workbench automatically splits the sequences up into smaller subsets and sends one subset at the time to NCBI. This is to avoid exceeding any internal limits the NCBI places on the number of sequences that can be submitted to them for BLAST searching. The size of the subset created in the CLC software depends both on the number and size of the sequences.
To start a BLAST job to search your sequences against databases held at the NCBI, go to:
Toolbox | BLAST (![]() )| BLAST at NCBI (
)| BLAST at NCBI (![]() )
)
Alternatively, use the keyboard shortcut: Ctrl+Shift+B for Windows
and ![]() +Shift+B on Mac OS.
+Shift+B on Mac OS.
This opens the dialog seen in figure 13.2
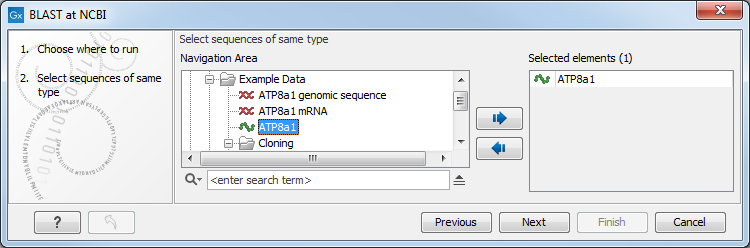
Figure 13.2: Choose one or more sequences to conduct a BLAST search with.
Select one or more sequences of the same type (either DNA or protein) and click Next.
In this dialog, you choose which type of BLAST search to conduct, and which database to search against (figure 13.3). The databases at the NCBI listed in the dropdown box will correspond to the query sequence type you have, DNA or protein, and the type of blast search you can chose among to run. A complete list of these databases can be found in BLAST databases. Here you can also read how to add additional databases available the NCBI to the list provided in the dropdown menu.
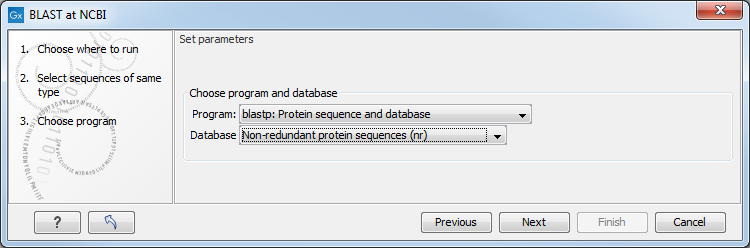
Figure 13.3: Choose a BLAST Program and a database for the search.
BLAST programs for DNA query sequences:
- blastn: DNA sequence against a DNA database. Searches for DNA sequences with homologous regions to your nucleotide query sequence.
- blastx: Translated DNA sequence against a Protein database. Automatic translation of your DNA query sequence in six frames; these translated sequences are then used to search a protein database.
- tblastx: Translated DNA sequence against a Translated DNA database. Automatic translation of your DNA query sequence and the DNA database, in six frames. The resulting peptide query sequences are used to search the resulting peptide database. Note that this type of search is computationally intensive.
BLAST programs for protein query sequences:
- blastp: Protein sequence against Protein database. Used to look for peptide sequences with homologous regions to your peptide query sequence.
- tblastn: Protein sequence against Translated DNA database. Peptide query sequences are searched against an automatically translated, in six frames, DNA database.
If you search against the Protein Data Bank protein database homologous sequences are found to the query sequence, these can be downloaded and opened with the 3D view.
Click Next.
This window, see figure 13.4, allows you to choose parameters to tune your BLAST search, to meet your requirements.
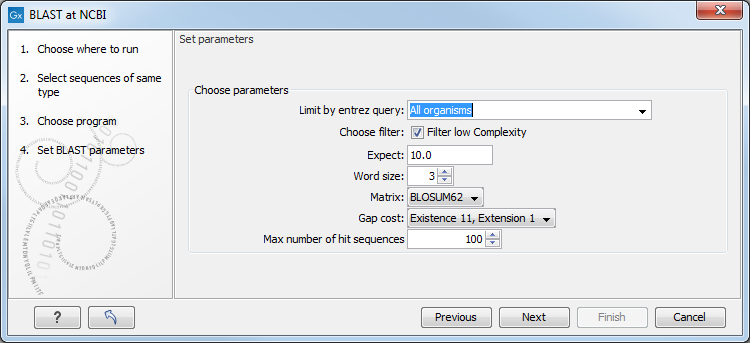
Figure 13.4: Parameters that can be set before submitting a BLAST search.
When choosing blastx or tblastx to conduct a search, you get the option of selecting a translation table for the genetic code. The standard genetic code is set as default. This setting is particularly useful when working with organisms or organelles that have a genetic code different from the standard genetic code.
The following description of BLAST search parameters is based on information from http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml.
- Limit by Entrez query. BLAST searches can be limited to the results of an Entrez query against the database chosen. This can be used to limit searches to subsets of entries in the BLAST databases. Any terms can be entered that would normally be allowed in an Entrez search session. More information about Entrez queries can be found at http://www.ncbi.nlm.nih.gov/books/NBK3837/#EntrezHelp.Entrez_Searching_Options. The syntax described there is the same as would be accepted in the CLC interface. Some commonly used Entrez queries are pre-entered and can be chosen in the drop down menu.
- Mask low complexity regions. Mask off segments of the query sequence that have low compositional complexity.
- Mask low complexity regions. Mask off segments of the query sequence that have low compositional complexity. Filtering can eliminate statistically significant, but biologically uninteresting reports from the BLAST output (e.g. hits against common acidic-, basic- or proline-rich regions), leaving the more biologically interesting regions of the query sequence available for specific matching against database sequences.
- Expect. The threshold for reporting matches against database sequences: the default value is 10, meaning that under the circumstances of this search, 10 matches are expected to be found merely by chance according to the stochastic model of Karlin and Altschul (1990). Details of how E-values are calculated can be found at the NCBI: http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html If the E-value ascribed to a match is greater than the EXPECT threshold, the match will not be reported. Lower EXPECT thresholds are more stringent, leading to fewer chance matches being reported. Increasing the threshold results in more matches being reported, but many may just matching by chance, not due to any biological similarity. Values of E less than one can be entered as decimals, or in scientific notiation. For example, 0.001, 1e-3 and 10e-4 would be equivalent and acceptable values.
- Word Size. BLAST is a heuristic that works by finding word-matches between the query and database sequences. You may think of this process as finding "hot-spots" that BLAST can then use to initiate extensions that might lead to full-blown alignments. For nucleotide-nucleotide searches (i.e. "BLASTn") an exact match of the entire word is required before an extension is initiated, so that you normally regulate the sensitivity and speed of the search by increasing or decreasing the wordsize. For other BLAST searches non-exact word matches are taken into account based upon the similarity between words. The amount of similarity can be varied so that you normally uses just the wordsizes 2 and 3 for these searches.
- Match/mismatch. A key element in evaluating the quality of a pairwise sequence alignment is the "substitution matrix", which assigns a score for aligning any possible pair of residues. The matrix used in a BLAST search can be changed depending on the type of sequences you are searching with (see the BLAST Frequently Asked Questions). Only applicable for protein sequences or translated DNA sequences.
- Gap Cost. The pull down menu shows the Gap Costs (Penalty to open Gap and penalty to extend Gap). Increasing the Gap Costs and Lambda ratio will result in alignments which decrease the number of Gaps introduced.
- Max number of hit sequences. The maximum number of database sequences, where BLAST found matches to your query sequence, to be included in the BLAST report.
The parameters you choose will affect how long BLAST takes to run. A search of a small database, requesting only hits that meet stringent criteria will generally be quite quick. Searching large databases, or allowing for very remote matches, will of course take longer.
Click Finish to start the tool.
BLAST a partial sequence against NCBI
You can search a database using only a part of a sequence directly from the sequence view:
select the sequence region to send to BLAST |
right-click the selection | BLAST Selection Against
NCBI (![]() )
)
This will go directly to the dialog shown in figure 13.3 and the rest of the options are the
same as when performing a BLAST search with a full sequence.