Demultiplexing output options
In the last wizard step, where you can specify the output options. If you choose to keep the default settings, three different types of output will be generated; 1) The demultiplexed reads, one output for each specified barcode (the file name starts with the sample name, followed by the specified barcode name), 2) The discarded reads that did not have a barcode (specified by the suffix "Not grouped"), and 3) a "Demultiplex Reads report", which shows the fraction of reads with and without a barcode (see figure 25.26).
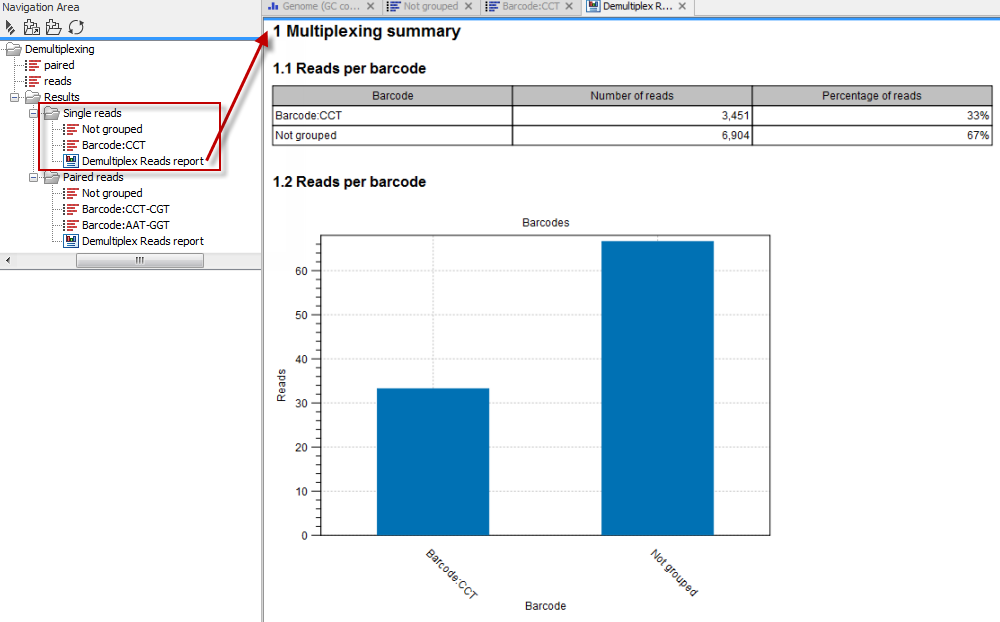
Figure 25.26: Three different outputs are generated when analyzing single reads with only one sample using the default output settings. If several samples had been mixed together there would be a sequence list for each sample (each specified barcode). The Demultiplex Reads report is shown in the right-hand side of the figure.
There is also an option to create subfolders for each sequence list. This can be handy when the results need to be processed in batch mode.
A new sequence list will be generated for each barcode containing all the sequences where this barcode is identified. Both the linker and barcode sequences are removed from each of the sequences in the list, so that only the target sequence remains. This means that you can continue the analysis by doing trimming or mapping. Note that you have to perform separate mappings for each sequence list.
An example of the demultiplexing summary report is shown in figure 25.27.
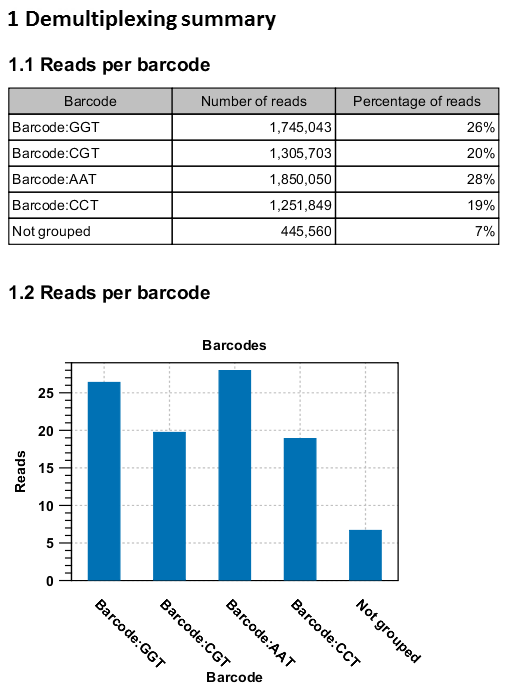
Figure 25.27: An example of a report showing the number of reads in each group. In this example four different barcodes were used to separate four different samples.