Run the Identify Known Mutations from Sample Mappings tool
To run the "Identify Known Mutations from Sample Mappings" tool go to the toolbox:
Toolbox | Resequencing Analysis (![]() ) |
Identify Known Mutations from Sample Mappings (
) |
Identify Known Mutations from Sample Mappings (![]() )
)
This opens the wizard shown where you can specify the read mapping(s) to analyze. Click Next to get the following options (figure 28.34):
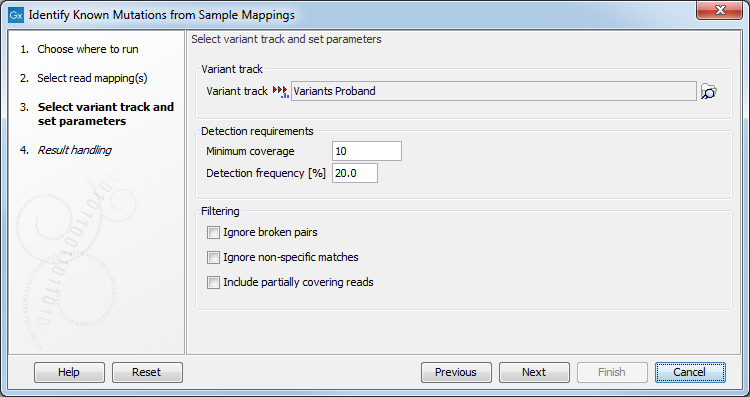
Figure 28.34: Select the variant track with the variants that you wish to use for variant testing.
- Variant track
-
- Variant track Select the variant track that contains the specific variants that you wish to test for in your read mapping. Note! You can only select one variant track at the time. If you wish to compare with more than one variant track, you must run the analysis with each individual variant track at the time.
- Detection requirements
-
- Minimum coverage The minimum number of reads that covers the position of the variant, which is required to set "Sufficient Coverage" to YES.
- Detection frequency The minimum allele frequency that is required to annotate a variant as being present in the sample. The same threshold will also be used to determine if a variant is homozygous or heterozygous. In case the most frequent alternative allele at the position of the considered variant has a frequency of less than this value, the zygosity of the considered variant will be reported as being homozygous.
- Filtering
-
- Ignore broken pairs When ticked, reads from broken pairs are ignored. Broken pairs may arise for a number of reasons, one being erroneous mapping of the reads. In general, variants based on broken pair reads are likely to be less reliable, so ignoring them may reduce the number of spurious variants called. However, broken pairs may also arise for biological reasons (e.g. due to structural variants) and if they are ignored some true variants may go undetected.
- Ignore non-specific matches Reads that have an equally good match elsewhere on the reference genome (these reads are colored yellow in the mapping view) can be ignored in the analysis. Whether you include these reads or not will be a tradeoff between sensitivity and specificity. Including them may lead to the prediction of transcripts that are not correct, whereas excluding them may mean that you will loose some true transcripts.
- Include partially covering reads Reads that partially overlap variants (see the blue box below for a definition) will be considered to enable the detection of variants that are longer than the reads. When the "Include partially covering reads" option is disabled, only fully covering reads will be counted for all annotations. Enabling the "Include partially covering reads" option means that all fully covering reads will be counted for all annotations, and that additionally, partially covering reads will be included in relevant annotations including Coverage. Thus, if a partial read is compatible with multiple variants in the same region, the sum of all Counts for that region may be greater than the Coverage, and the sum of all Frequencies for that region may be higher than 100%.
|
A fully covering read is described as such:
|
Click Next to go to the next wizard step (figure 28.35). At this step the output options can be adjusted.
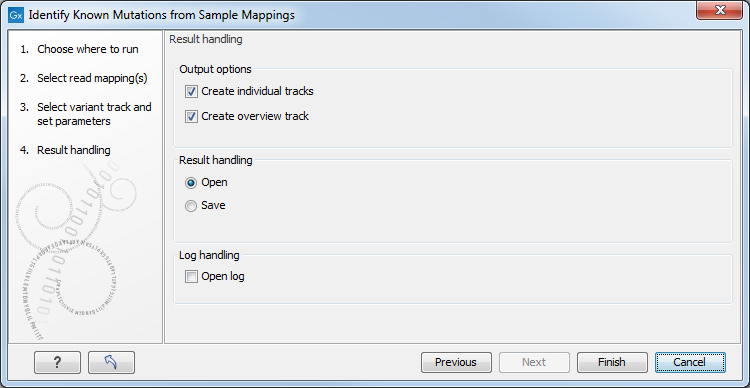
Figure 28.35: Select the desired output format(s). If using the default settings, two types of output will be generated; individual tracks and overview tracks.
The output options are:
- Create individual track For each read mapping an individual track is created with the observed frequency, average base quality, forward/reverse read balance, zygosity and observed allele count.
- Create overview track The overview track is a summary for all samples with information about whether the coverage is sufficient at a given variant position and if the variant has been detected; the frequency of the variant.
Specify where to save the results and click on the button labeled Finish.