Analyze QIAseq xHYB Viral Panel Data
The Analyze QIAseq xHYB Viral Panel Data template workflow trims reads, performs taxonomic profiling, and calls viral variants.
It is suitable for data generated with the QIAseq xHYB viral panels:
- QIAseq xHYB Respiratory Panel
- QIAseq xHYB Viral STI Panel
- QIAseq xHYB Adventitious Agent Panel
- QIAseq xHYB MPXV Panel
QIAGEN reference data set
The QIAseq xHYB Viral Panels reference data set is available from QIAGEN Sets Reference Data Library accessible via References (![]() ) in the top Toolbar.
) in the top Toolbar.
Launching the workflow
The Analyze QIAseq xHYB Viral Panel Data workflow is at:
Toolbox | Template Workflows (![]() ) | Microbial Workflows (
) | Microbial Workflows (![]() ) | QIAseq Analysis (
) | QIAseq Analysis (![]() ) | Analyze QIAseq xHYB Viral Panel Data (
) | Analyze QIAseq xHYB Viral Panel Data (![]() )
)
Launch the workflow and step through the wizard.
- Select the sequence list(s) containing the reads to analyze. Click on Next.
- Select a reference data set or select "Use the default reference data" to configure the reference data elements individually in subsequent wizard steps (figure 3.79). Click on Next.
- Choose whether batch units should be defined based on organization of the input data, or by provided metadata (figure 3.80). For information on how to use metadata when running part of a workflow multiple times, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Running_part_workflow_multiple_times.html.
- Next, you can review the batch units resulting from your selections above. Click on Next.
- Verify or select the viral taxonomic profiling index (figure 3.81) and click on Next.
- Verify or select the host taxonomic profiling index and click on Next.
- Select the viral reference database(s). If in the first step you selected the QIAseq xHYB Viral Panels reference set, you can now select which of the available viral reference databases from that set to apply (3.82). If you chose to use the default reference data, select a reference database and click on Next.
- Verify or select the control genes and click on Next.
- Specify the trim settings.
- Specify Taxonomic Profiling settings (figure 3.83).
- Specify Low Frequency Variant Detection settings, see figure 3.84.
- Finally, select a location to save outputs to and click on Finish.
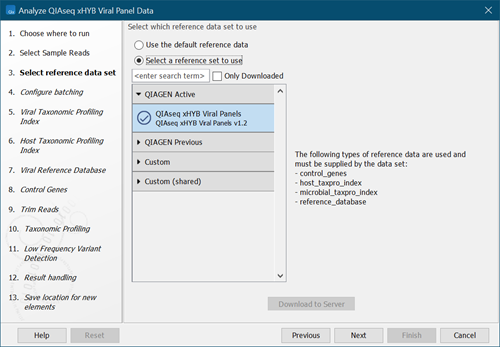
Figure 3.79:
Select reference data set.
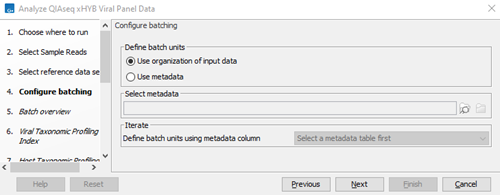
Figure 3.80:
Define batch units.
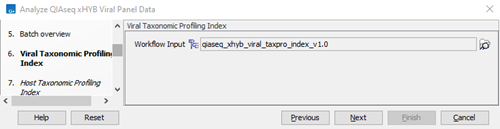
Figure 3.81:
Select viral taxonomic profiling index.
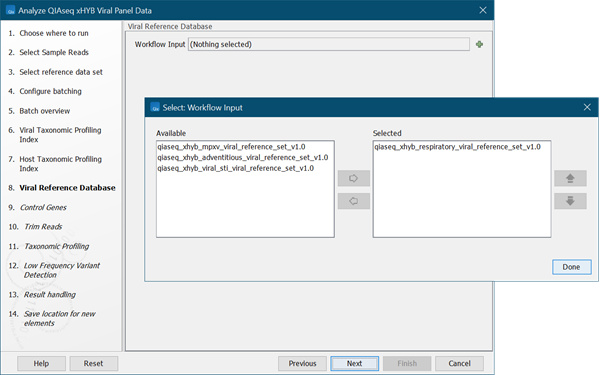
Figure 3.82:
Select one or more viral reference databases.
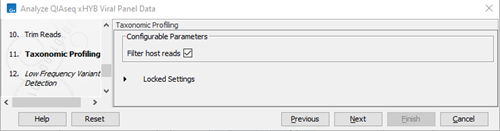
Figure 3.83:
Set taxonomic profiling parameters.
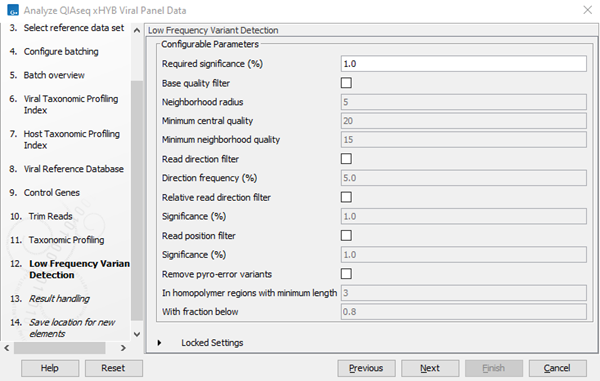
Figure 3.84:
Low frequency variant detection settings.
Tools in the workflow and outputs generated
The workflow Analyze QIAseq xHYB Viral Panel Data consists of the below mentioned tools. See figure 3.85 for a full overview of the workflow.
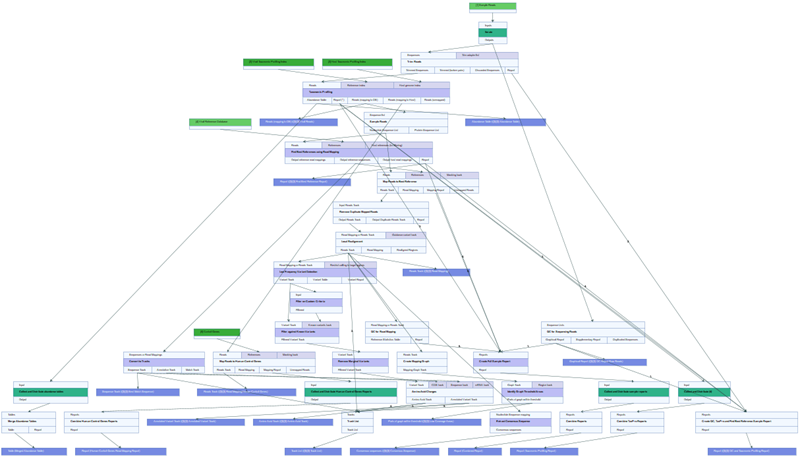
Figure 3.85:
The Analyze QIAseq xHYB Viral Panel Data workflow layout.
The tools used in this workflow are:
- QC for Sequencing Reads performs basic QC on the sequencing reads. The output, which here is included in a combined report, can be used to evaluate the quality of the sequencing reads. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=QC_Sequencing_Reads.html.
- Trim Reads removes adapter sequences and low quality nucleotides. The appropriate settings for the Trim Reads tool depends on the protocol used to generate the reads.
See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Trim_Reads.html.
- Taxonomic Profiling provides insight into the taxonomic composition of the sample and estimates the relative abundance of the detected taxa. See Taxomonic Profiling. Host reads, i.e. reads that map to the host taxonomic profiling index, do not count toward the taxonomic profiling result, but are used as input for Map Reads to Human Control Genes. Viral reads - reads that map to the viral taxonomic profiling index - are later used as input for Find Best References using Read Mapping.
- Map Reads to Human Control Genes maps the host reads output from Taxonomic Profiling to the host taxonomic profiling index, to a reference of human control genes. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Map_Reads_Reference.html. This serves as a QC step to verify mapping to the human control genes.
- Find Best References using Read Mapping maps the viral reads output from Taxonomic Profiling to the selected viral reference database to identify which reference sequence is the "Best match". See Find Best Reference using Read Mapping.
- Map Reads to Best Reference maps viral reads to the identified "Best match" reference. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Map_Reads_Reference.html. The output reads track is used as input for Remove Duplicate Mapped Reads.
- Remove Duplicate Mapped Reads removes duplicate reads derived from PCR amplification (or other enrichment) during sample preparation from the mapping. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Remove_Duplicate_Mapped_Reads.html. The output reads track is used as input for Local Realignment.
- Local Realignment improves the alignment of the reads in the reads track. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Local_Realignment.html.
- Low Frequency Variant Detection calls variants in the read mapping that are present at low frequencies. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Low_Frequency_Variant_Detection.html.
- Filter on Custom Criteria, Filter against Known Variants, and Remove Marginal Variants together remove variants that fall below a set of thresholds. For this workflow for instance, coverage >30 and frequency >20% is required. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Variant_filtering.html.
- Amino Acid Changes use the called variants to generate a track of amino acid changes. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Amino_Acid_Changes.html.
- Create Mapping Graph and Identify Graph Threshold Areas combined creates a track with regions with coverage below a threshold. For this workflow, the threshold is set to 30. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Create_Mapping_Graph.html and http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Identify_Graph_Threshold_Areas.html.
- Extract Consensus Sequence makes a consensus sequence from the read tracks from Local Realignment. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Extract_Consensus_Sequence.html.
- QC for Read Mapping performs QC of the read mapping. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=QC_Read_Mapping.html.
The sample-specific outputs provided by this workflow are:
- QC Report Raw Reads provides QC metrics on the raw reads.
- Abundance Table provides the abundance of each identified taxa, along with their full taxonomy. See Taxomonic Profiling Abundance.
- Read Mapping Human Control Genes contains the host reads mapped against the control gene reference.
- Viral Reads is the list of reads that mapped to the viral taxonomic profiling index.
- Find Best Reference Report is a report of the references found using Find Best Reference.
- Best Match Sequence the "Best match" reference sequence as identified by the Find Best References using Read Mapping tool.
- Read Mapping displays the mapping of viral reads against the "Best match" viral reference.
- Consensus Sequence contains the viral consensus sequence(s) extracted from the Local Realignment reads track.
- Annotated Variant Track contains the list of detected variants left after filtering, annotated with amino acid changes.
- Amino Acid Track contains a list of amino acid changes.
- Low Coverage Areas provides a list of low coverage regions in the mapping of viral reads against the "Best match" viral reference.
- Track List is a collection of the following viral tracks: consensus sequence, reads, variants, amino acid changes, and low coverage regions.
- QC and Taxonomic Profiling Report combines QC Report Raw Reads and the Taxonomic Profiling report.
The combined outputs provided by this workflow are:
- Taxonomic Profiling Report combines taxonomic profiling report content across samples in the workflow run. See Taxomonic Profiling Report.
- Human Control Genes Read Mapping Report holds information about the mapping of host reads to the human control genes for all samples in the workflow run.
- Combined Report combines information from various tools, including QC, taxonomic profiling and mapping reports.
- Merged Abundance Table provides abundances for the detected taxa for all samples in the workflow run. See Taxomonic Profiling Abundance Table.