LightSpeed Long Reads to Germline Variants
The LightSpeed Long Reads to Germline Variants (beta) tool is designed to provide variant calls from Oxford Nanopore and PacBio HiFi sequencing data within a very short timeframe.
The tool can perform quality trimming, mapping, and phased germline variant calling. For a description of each step, see LightSpeed Methods.
Input reads must be demultiplexed and/or trimmed for adapter sequences as relevant. For data produced using QIAseq Long Read panels, a tool is available for trimming and demultiplexing, see LINK.
LightSpeed Long Reads to Germline Variants (beta) can only analyze one sample per analysis start. To analyze samples in batch, LightSpeed Long Reads to Germline Variants (beta) must be included in a workflow (see Batching). Template workflows for LightSpeed analyses are available (see Template Workflows), but it is also possible to create custom workflows. Read about workflows here https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Workflows.html.
To run the LightSpeed Long Reads to Germline Variants (beta) tool go to:
Tools | LightSpeed (![]() ) | LightSpeed Long Reads to Germline Variants (beta) (
) | LightSpeed Long Reads to Germline Variants (beta) (![]() )
)
If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
In the first wizard step, specify read files (fastq or unaligned BAM) and a reference sequence (figure 3.7):
- Input data
- Reads Fastq or unaligned BAM files for analysis.
- Sequencing platform The sequencing platform used to generate the reads.
- References
- References The reference sequence that reads will be mapped to.
- Reference masking
- No masking Reads are mapped to the full reference sequence.
- Exclude annotated Reads are mapped to the full reference sequence except regions specified in the masking track.
- Include annotated only Reads are only mapped to the regions specified in the masking track.
- Masking track The track specifying the masking regions.
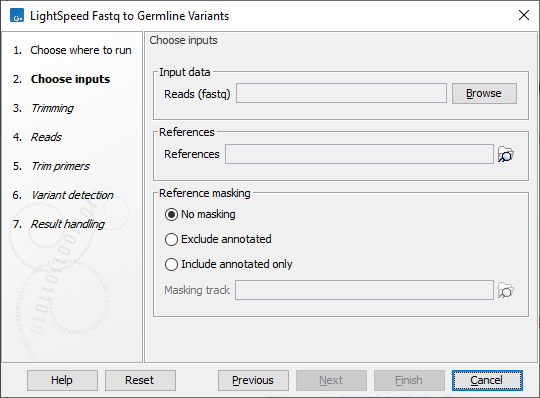
Figure 3.7: Input fastq files and references, and, optionally, a track for reference masking.
Next, options are available for quality trimming (figure 3.8):
- Quality trimming
- Quality trim Reads are trimmed for low quality nucleotides.
- Quality trim limit Adjust the quality trim limit for softer or harder trimming. Read more about the quality trim limit here: https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Quality_trimming.html.
- Minimum read length after quality trim Trimmed reads shorter than this length are removed.
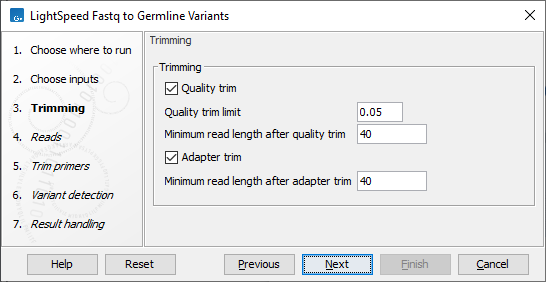
Figure 3.8: Options for trimming.
Next, options are available for variant detection (figure 3.9):
- Variant detection
- Ploidy The expected maximum number of different alleles.
- Restrict calling to target regions Optional: A track providing the regions to be inspected when calling variants. When no track is provided, all positions in the mapping are considered.
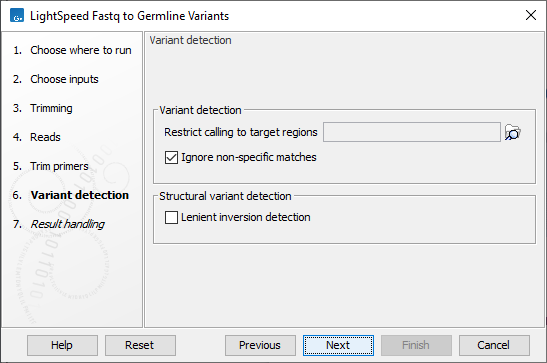
Figure 3.9: Options for variant detection.
Next, options are available for variant filtering (figure 3.10):
- Variant filters
- SNV minimum average quality The minimum average quality of detected SNVs and MNVs.
- Indel minimum average quality The minimum average quality of detected indels.
- Minimum QUAL The minimum required QUAL score for detected variants. The QUAL score reflects the likelihood of the variant being a real variant.
- Minimum allele count The minimum allele count for detected variants.
When set to 1, special handling is applied to variants that are only supported by 1 read and have a coverage of 1 or 2. These variants are not assessed for significance, but are reported when the following criteria are met:
- The average quality score is 35 or higher.
- The site has no overlapping non-specific reads.
- Minimum frequency (%) The minimum frequency for detected variants.
- STR annotations and filter Annotate and filter variants that change the number of repeats in short tandem repeat (STR) regions, excluding homopolymers.
- Minimum repeat count The minimum number of repeats, excluding the variant, that an allele must have to be annotated as an STR variant. For example if a reference allele "AGAGAGAG" is called, and a low frequent stutter insertion allele is called "AGAGAGAGAG", the repeat count is 4.
- Minimum repeat region length The minimum length of all repeats combined, excluding the variant, that an allele must have to be annotated as an STR variant. For example if a reference allele "AGAGAGAG" is called, and a low frequent stutter insertion allele "AGAGAGAGAG" is called, the repeat region length is 8.
- Maximum repeat element length The maximum length of a repeat unit, that an allele can have to be annotated as an STR variant. For example, for the dinucleotide repeat "AGAGAGAG", the repeat unit length is 2.
- STR filter Enable to remove variants annotated as STR "Yes" from the results, provided that they are below the frequency threshold defined under "Remove STR variants with frequency below".
- Remove STR variants with frequency below Specify the frequency threshold below which STR variants will be removed from the results.
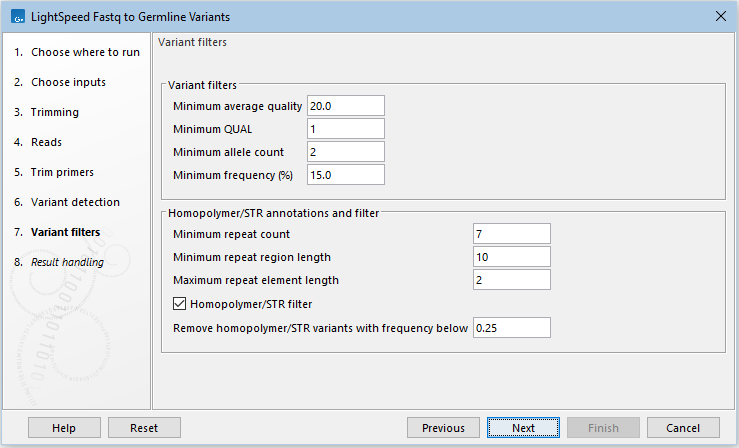
Figure 3.10: Options for variant filtering.
In the final wizard step, choose which outputs should be generated and whether results should be saved or opened. If a reads track is selected as output, runtime will increase.
Subsections