UMI grouping
All of the LightSpeed tools can group reads based on Unique Molecular Identifiers (UMIs). Both protocols where the UMI is present on only one read in a pair or both reads in a pair (duplex UMI) are supported.
The UMI sequence is recorded and removed from the reads before trimming and mapping, or it can be read from the fastq read header. After the reads have been mapped, reads with similar UMI sequence and mapping position are merged into a consensus UMI read.
For duplex UMIs, UMI grouping is a two step process, where reads are first grouped to simplex reads and then to duplex reads.
The consensus is calculated following these rules:
- At conflicting positions, the most common base is included in the consensus read.
- If the conflicting bases are equally represented the consensus can be generated in two ways:
- When one of the bases at the conflicting position is identical to the reference symbol, the reference symbol is included in the consensus read.
- When none of the bases at the conflicting position is identical to the reference symbol, an N is inserted in the consensus read.
Q-scores are assigned to the bases in the UMI read as follows:
- For UMI groups with only one read (singleton groups), the Q-scores of the bases in the original read are used.
- For UMI groups with more than one read, and where all reads agree on the base, the average Q-score of the bases is used. However, if this value is smaller than the adaptation of the MAGERI Q-score, Q_M (described below), the Q_M value is used.
- At conflicting positions, where there is a most common base, the Q-score assigned to a base in the consensus read is calculated using an adaptation of MAGERI (a method described in [Shugay et al., 2017]). Specifically, the Q_M value is used as outlined in the following definitions:
- count = number of reads with the winning nucleotide
- total = total number of reads in UMI group
- f = count/(total + 0.9)
- Q_M = 60/3*(4*f -1)
- If the conflicting bases are equally represented, the consensus base will either be the reference symbol or N. In both cases no Q-score assignment is made to the base.
Examples of the resulting UMI read Q-scores are given in figure 3.1.
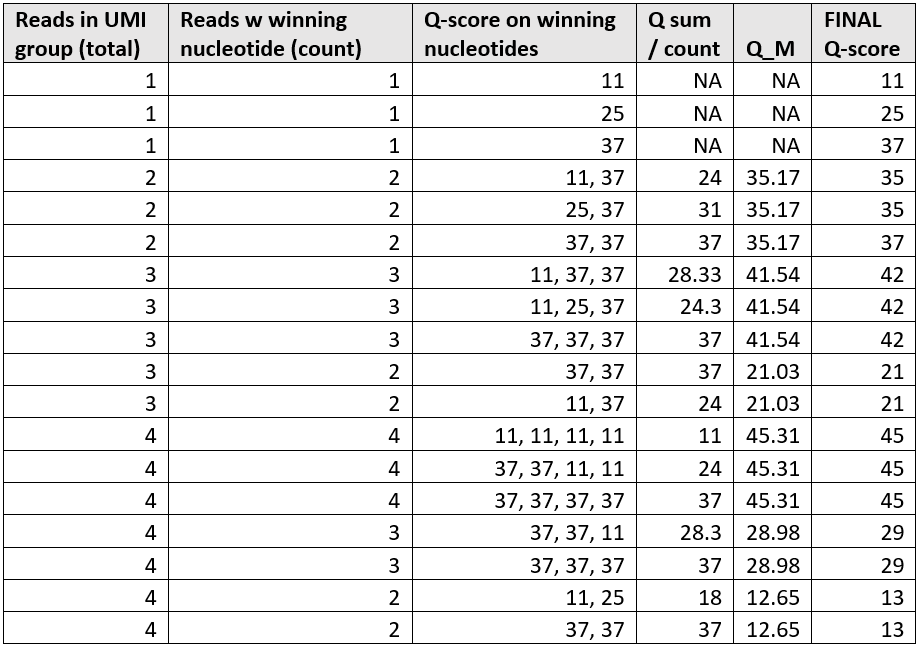
Figure 3.1: Assigned Q-scores exemplified for various UMI group sizes, base quality scores and base ambiguity among contributing reads.
When variants are called from UMI reads, additional UMI specific annotations are added, see Germline variant detection or Somatic variant detection.
For limitations in UMI grouping, see Limitations.
Subsections