Output from the Detect QIAseq Methylation workflow
The Detect QIAseq Methylation workflow generates the following outputs:
- A Track List that includes - among others - the following tracks:
- A read mapping
- A target coverage track
- A methylation levels track
- Several reports (see figure 16.2)
Figure 16.2: Reports generated by the workflow.
It is recommended to look at Section 3 of the Methylation report in order to check that there are no problems with the bisulfite conversion reaction (see figure 16.3). The CHG and CHH values can be used as a proxy for the efficiency of bisulfite conversion, as most methylation in human samples is seen in CpG contexts (except for certain cell types [He and Ecker, 2016]). Typically CHG and CHH will be <2%.
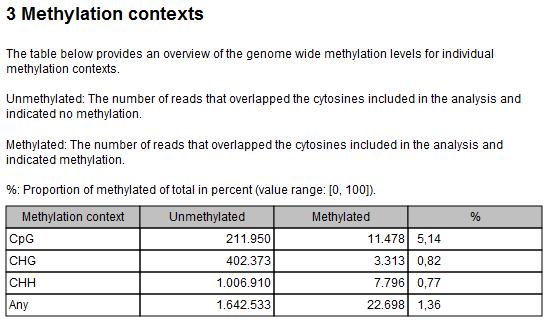
Figure 16.3: Methylation contexts table of the Methylation report.
Note that in Section 4 of the Methylation report, it is expected that most R1 related values are 0, as the primer is unaligned from the start of read 1, meaning that the first 25 bases are (almost) never aligned.
Also note that in the UMI Group Creation report, the last plot shows the "Nucleotide percentages of the unique molecular barcode symbols". This plot looks like the one defined as bad quality for Targeted DNA application (see Quality Control for the Identify QIAseq DNA Variants workflow). This is however expected in Targeted Methyl applications as the UMI for methylation is "NNCNNCNNCNN", meaning that red "C" bars follow a different distribution from the other nucleotides.
The Detect QIAseq Methylation with T Cell Infiltration workflow generates two additional outputs:
- A methylation profile region track
- A methylation profile report, showing the percentage of the sample estimated to come from epithelial cells (Epi), fibroblast cells (Fib), and immune cells (IC).
For more details on these additional outputs, see Predict Methylation Profile.
|
The provided methylation reference database (Homo_sapiens_CpG_hg19_EpiFibIC) is constructed from a limited set of epithelial, fibroblast, and immune cell types. It is possible that the epithelial, fibroblast, and immune cell types in any given sample differ from these, and have different methylation profiles. If this is the case, then prediction performance may suffer.
We advise testing the database on pure samples and mixtures with known proportions to see if it is suitable for the analysis.
If it is necessary to create a database, use the tool described in Create Methylation Database. |