Detect MSI Status with Baseline Creation
The Detect MSI Status with Baseline Creation template workflow is designed to support the QIAseq MSI booster panel, SDHS-10101-11981Z. It can be used with either the hg19 or hg38 reference genomes.
To run the workflow, go to:
Template Workflows | Biomedical Workflows (![]() ) | QIAseq Sample Analysis (
) | QIAseq Sample Analysis (![]() ) | QIAseq DNA Workflows (
) | QIAseq DNA Workflows (![]() ) | Detect MSI Status with Baseline Creation (
) | Detect MSI Status with Baseline Creation (![]() )
)
In the first dialog select the reads for analysis (figure 14.5).
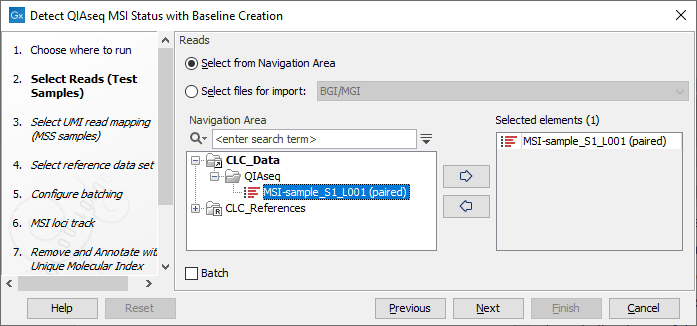
Figure 14.5: Select the sequencing reads by double-clicking on the file name or by clicking once on the file name and then on the arrow pointing to the right hand side.
In the next dialog, select the UMI read mappings of the MSS samples forming the Loci Baseline (figure 14.6). Baseline mappings should be created from at least 15 MSS FFPE samples, preferably samples analyzed on the same sequencing instrument. Ideally, the samples would be from normal biopsies matched with the tumor samples being analyzed. By including the normal sample in the analysis, bias can be avoided.
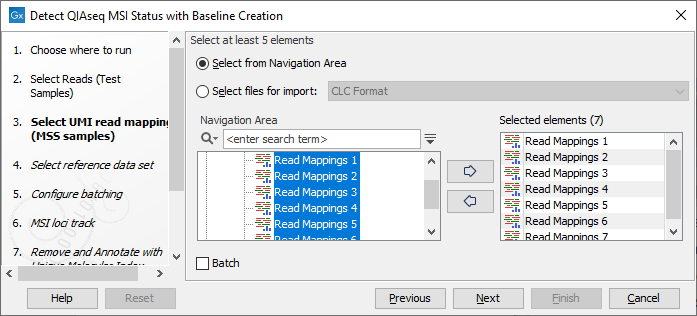
Figure 14.6: Select at least 15 MSS samples to form a baseline.
If you already have read mappings for the MSS samples, these can be used as input to this workflow. You may have such mappings if the MSS samples have been analyzed together with data from other QIAseq custom panels or from a QIAseq Targeted DNA panel.
If you do not already have read mappings for the MSS samples then these can be generated using an edited copy of the Detect MSI Status with Baseline Creation workflow. Modify a copy of this workflow by right clicking on the workflow in the Toolbox and selecting the menu option "Open Copy of Workflow". Delete the MSI tools Detect MSI Status and Generate MSI Baseline from the workflow, save it, and then run it. Provide the MSS samples as input. The output read mappings from this workflow run can then be used as inputs to the original Detect MSI Status with Baseline Creation workflow.
In the next dialog, choose either the QIAseq TMB Panels hg38 (no alternative analysis set) or a custom reference data set based on QIAseq DNA Panel hg19. Note that your reference data must match the reference sequence used for your MSS UMI read mappings.
|
To create a reference data set based on QIAseq DNA Panel hg19, first download the MSI Loci Track in hg19 coordinates. This is available as a Reference Data Element that can be found under the "QIAGEN Sets" tab of the Reference Data Manager. Then create a Custom Reference Data Set to match this workflow as described here http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Custom_Sets.html. |
In the Configure batching and Batch overview wizard steps use the default settings. The MSI Loci Track step can be configured to use either the 9 loci or the 27 loci version of the loci (figure 14.6), or a custom selection if this has been created beforehand (for more on how to create a custom selection of loci see Detect MSI Status).
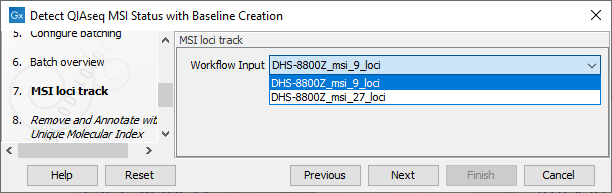
Figure 14.7: Select an MSI loci track.
In the Map Reads to Reference dialog, it is possible to configure masking. A custom masking track can be used, but by default, the masking track is set to GenomeReferenceConsortium_masking_hg38_no_alt_analysis_set, containing the regions defined by the Genome Reference Consortium, which serve primarily to remove false duplications, including one affecting the gene U2AF1. Changing the masking mode from "No masking" to "Exclude annotated" excludes these regions.
Select settings for Result handling and a Save location for new elements and press Finish to start the analysis.