Annotate RNA Variants
The Annotate RNA Variants tool annotates variants likely to arise from alternative splicing rather than DNA changes. The annotations are useful for downstream filtering.
Three classes of annotations are added "known introns", "splice variants", and "closest exon". These are described briefly below.
Known introns annotation
When RNA and DNA reads are sequenced together "index-hopping" can occur, which leads to small numbers of RNA reads being found in the DNA file. If these RNA reads splice across a short intron they can be interpreted as providing support for one or more deletions spanning the intron. An example of this is shown in figure 11.1. The Annotate RNA Variants tool slides each of these deletions independently to the left and right to see if they can be aligned with an exon boundary without changing the deleted sequence. If this is possible, the deletion is annotated with "Matches known intron = Yes".

Figure 11.1: Example of two variants that will be annotated as matching a known intron.
Splice variants annotations
Some variants likely to arise from alternative splicing rather than DNA changes. Although these are often due to tandem acceptor splice variants not present in the mRNA annotations, they can also be clinically relevant. A simple example unlikely to be of clinical relevance is shown in figure 11.2.
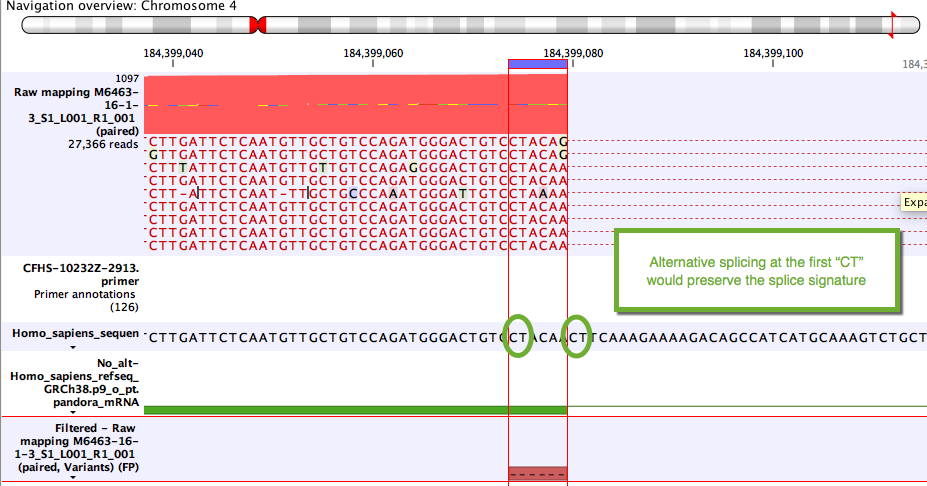
Figure 11.2: Example of a variant that will be annotated as a splice variant conserving the splice signature.
To detect such variants, the tool lists all variants within 30nt of each intron. All combinations of these variants are then generated such that for input variants v1, v2, v3, the output combinations would be: [v1], [v2], [v3], [v1,v2], [v1,v3], [v2,v3], [v1,v2,v3]. At most 31 combinations of variants are generated in this way as the number of possible combinations of variants quickly becomes unmanageable. Combinations of variants that are incompatible are not generated. For example if v1 and v2 are overlapping deletions, then combinations [v1,v2] or [v1,v2,v3] are not generated as these are not consistent with a single RNA molecule.
For each combination of variants, the DNA (extracted from the genome) is aligned to the variants + RNA (extracted from mRNA annotations). If the alignment can be made without mismatches, and with only a single gap, then the variants can be explained by a new intron with the coordinates of the gap. All such variants are then annotated with "Possible splice variant = Yes". Additional annotations are added based on the splice signature of the new intron:
- "Conserved splice signatures" with values "Yes" or "No" according to whether the splice signature matches that of the original intron.
- "Canonical splice signature" and zero or more comma-separated values from the list "GT-AG", "GC-AG", "AT-AC"
- "Possible splice signatures" and one or more comma-separated values from the list "AA-AA", "AA-AC" ... "TT-TT"
A variant may be investigated multiple times if it is close to multiple introns. If this happens, the annotations are updated such that "Conserved splice signature" may change from "No" to "Yes", and splice signatures may be added to the "Canonical splice signature" and "Possible splice signatures" lists.
Closest exon annotations
A common error mode of RNA-Seq variant calling is when a variant lies within an intron close to an exon. In this case the frequency of the variant may be artificially inflated if reads without the variant are spliced and have no coverage in the intron.An example is shown in figure 11.3. Here the coverage is 210 at the position where the SNV is called, but >2500 in the adjoining exon. The "real" variant frequency is closer to 16/2500 < 1% than 16/210 > 5%.
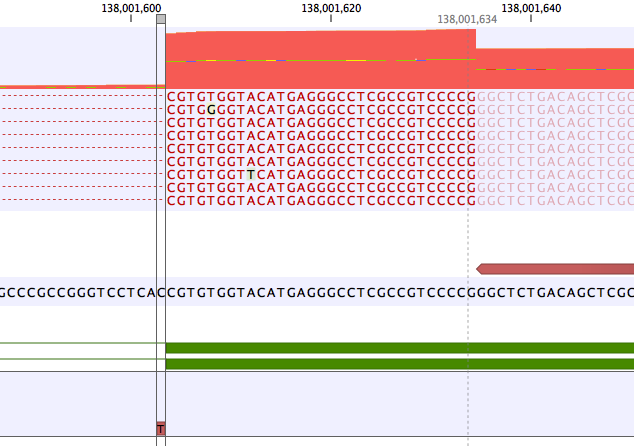
Figure 11.3: Example of a variant that is called with frequency >5%, but whose frequency corrected to the coverage at the nearby exon is very low.
The tool adds an annotation "Distance to nearest exon" to all non-reference variants within 100 nt of an exon. The nearest exon is that which returns the smallest positive distance among the following four options: (exonStart - variantStart, exonStart - variantEnd, variantStart-exonEnd, variantEnd-exonEnd).
If a read mapping is provided, the tool also annotates variants with the "Frequency compared to closest exon". This frequency is 100 * count / coverage at nearest exon. The coverage at the nearest exon is actually the coverage at the boundary of the exon. Coverage is calculated per match rather than per read i.e. broken pairs count as 2, pairs count as 1, and single reads count as 1. Coverage also includes ambiguously mapped reads.
In all cases where count > coverage at nearest exon, the tool reports "Frequency compared to closest exon = 100%" which in practice means that this annotation cannot be used to filter the variant. This can happen for a variety of reasons. For example:
- The nearest exon has no coverage - perhaps an exon with a boundary slightly further away from the variant has all the coverage
- Primers positioned in the intron generate more reads than primers in the adjacent exon
The Annotate RNA Variants tool can be found in the Toolbox at:
Toolbox | Resequencing Analysis (![]() ) | Variant Annotation (
) | Variant Annotation (![]() ) | Annotate RNA Variants (
) | Annotate RNA Variants (![]() )
)
The tool takes a Variant Track as input (figure 11.4)
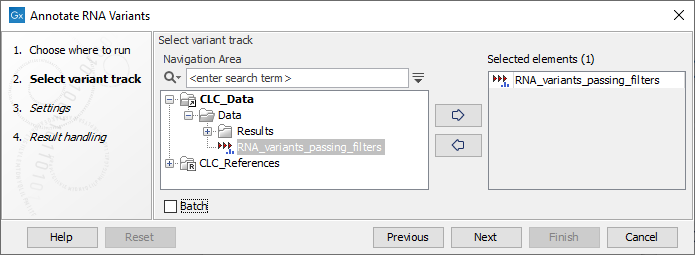
Figure 11.4: Select a variant track.
In the next dialog figure there are two parameters to set (figure 11.5)
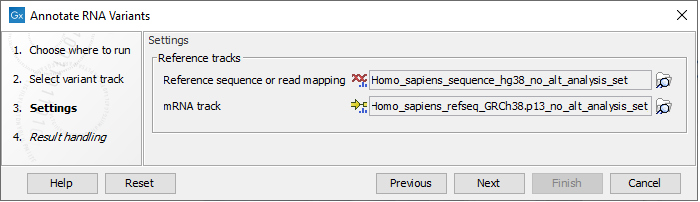
Figure 11.5: Select a reference genome or read mapping, and an mRNA track.
- Reference sequence or read mapping - variants will only be annotated with the coverage at the nearest exon if a read mapping is provided.
- mRNA track
The tool outputs an annotated Variant Track.
Subsections