Convert Annotation Track Coordinates
Convert Annotation Track Coordinates converts annotation coordinates, either from hg19 coordinates to hg38 coordinates, or vice versa. This remapping of coordinates, also referred to as 'liftover', makes use of the NCBI Remapping Service. Network access is therefore necessary to use this tool.
Note that the tool has the limitation that it only works for tracks with less than 30,000 annotations. If you have tracks with 30,000 annotations or more you need to create subtracks, run the tool on the subtracks and merge the resulting tracks. In addition, regions with no strand annotation, will be annotated to the negative strand after the conversion.
This tools uses assembly GCF_000001405.26 for hg38 and assembly GCF_000001405.13 for hg19.
To run Convert Annotation Coordinates Track, go to:
Toolbox | Biomedical Genomics Analysis (![]() ) | Biomedical Utility Tools (
) | Biomedical Utility Tools (![]() ) | Convert Annotation Track Coordinates (
) | Convert Annotation Track Coordinates (![]() )
)
The tool takes annotation tracks, typically primer tracks or target region tracks, as input (see figure 6.17).
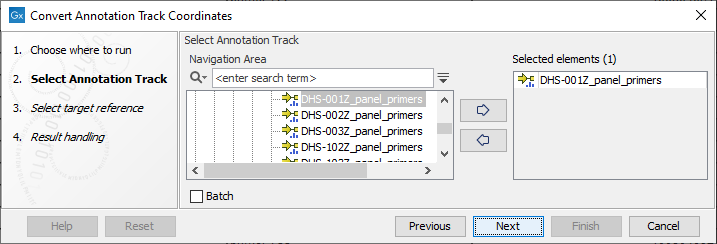
Figure 6.17: Selection of a primer annotation track with hg19 coordinates for coversion to hg38 coordinates.
In the next step, a target reference must be selected (see figure 6.18). If the annotation track used as input was based on an hg19 reference, select an hg38 reference genome. If the annotation track used as input was based on an hg38 reference, select an hg19 reference genome. If not already available, a copy of the relevant target reference can be downloaded using the Reference Data Manager.
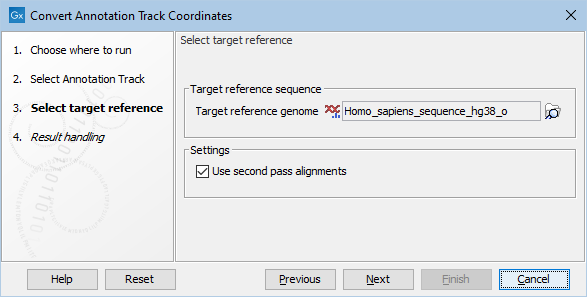
Figure 6.18: This dialog allows selection of the reference sequence you would like to lift over to. Under settings you can choose to use the second pass alignments option.
Under settings you can choose to use the Use second pass alignments option that allows alignment to duplicated sequences. For more information about second pass alignment, please see https://www.ncbi.nlm.nih.gov/genome/tools/remap/docs/whatis and https://www.ncbi.nlm.nih.gov/genome/tools/remap/docs/alignments.
This tool outputs an annotation track with coordinates based on the target reference and a report. If the remapping was done without selecting the Use second pass alignments option, the report will typically list the potentially skipped intervals, that is, the annotations that could not be mapped to coordinates of the selected target reference.
If remapping was done with the Use second pass alignment option selected, the report lists Skipped intervals, Annotations with multiple intervals that were split because not all intervals could be remapped and Duplicated intervals. The duplicated intervals table lists multiple outputs for the same input, which means that the report will have multiple entries for the same chromosome, name, and interval as shown in figure 6.19.
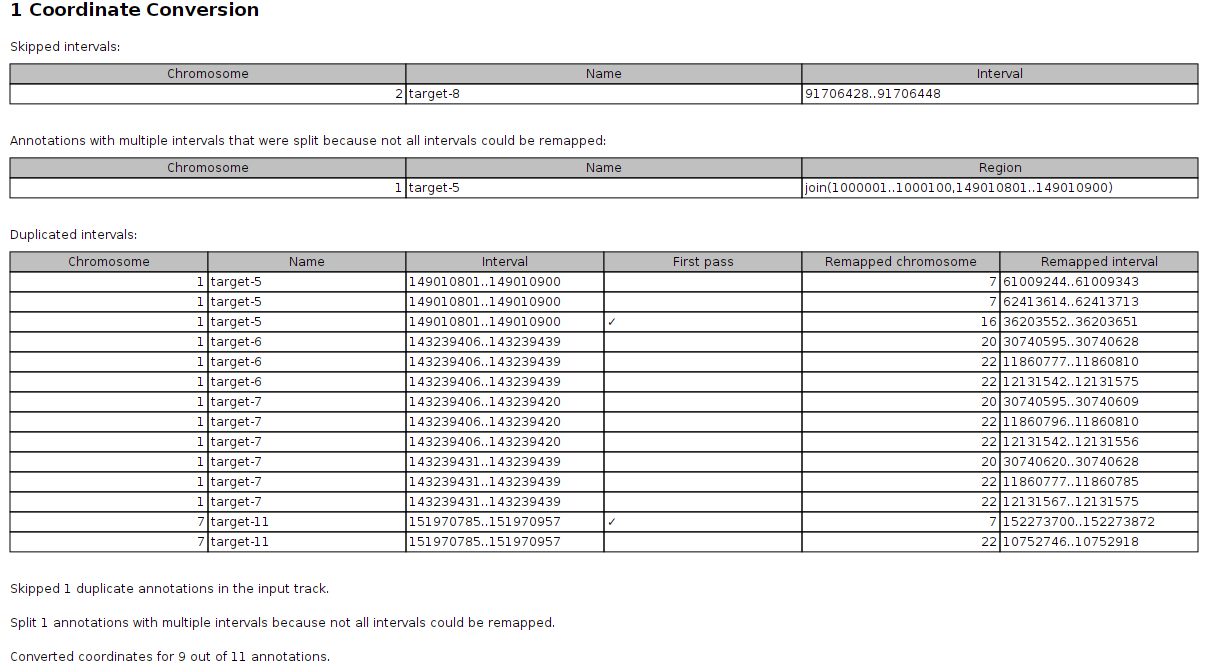
Figure 6.19: The Convert Annotation Track Coordinates report also lists duplicated annotations.
If multiple outputs were generated for some of the annotations, it possible to manipulate the resulting Annotation Track. This can be done by opening the output file with the extension (Converted) in table view and then choosing the subset of the output annotations track that is of relevance by selecting the relevant rows and clicking on the button labeled Create Track from Selection.
Input annotations with intervals defining a region between positions (similar to insertions) are converted to one base intervals covering the position after the input interval. This one base interval is then converted to new track coordinates. And then made back into an interval defining the region between the converted position and the one before it.