Immune Repertoire Analysis
Using RNA-Seq data as input, the Immune Repertoire Analysis tool can be used to characterize either the T or B cell receptor repertoire.
The tool requires a reference data sequence list (![]() ) containing reference sequences for the V, D, J and C segments.
) containing reference sequences for the V, D, J and C segments.
Whether the tool identifies T or B cell receptors depends on the types of reference segments present in the provided sequence list. The tool does not accept sequence lists containing reference sequences for both TCR and BCR.
The Reference Data Manager (see Reference Data Management) offers two QIAGEN sets for this tool. Each set contains a sequences list for Immune Repertoire Analysis:
- QIAseq Immune Repertoire Analysis for analysis of TCR human data.
- QIAseq Immune Repertoire Analysis Mouse for analysis of TCR mouse data.
If reference data is needed for BCR or for a different species than those above, Import Immune Reference Segments can be used to import reference data, see Import Immune Reference Segments.
|
The tool assumes that one read spans all segment types (V, D, J and C) in order to successfully report the clonotype. It is therefore recommended to collapse overlapping paired-end reads using Merge Overlapping Pairs, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Merge_Overlapping_Pairs.html. |
Identification of clonotypes
Clonotyping a read consists of identifying which V, D, J and C segments from the reference data are used and extracting the CDR3 region found between the conserved amino acids.
V and C segments are rather long (![]() bp), whereas J segments are relatively short (
bp), whereas J segments are relatively short (
![]() bp) and D segments are even shorter (
bp) and D segments are even shorter (
![]() bp).
The segments identification is therefore performed using different strategies.
bp).
The segments identification is therefore performed using different strategies.
First, the tool identifies the V and J segments.
For V segments, the Map Reads to Reference tool is used internally.
For the J segment, a strategy similar to IMSEQ [Kuchenbecker et al., 2015] is used. First, a pairwise alignment with a 15 bp subsequence of the full segment called a Segment Core Fragment (SCF) is performed to find candidates for full pairwise alignments. If the pairwise alignment of an SCF to the read has a sufficiently small number of errors, it is nominated as a candidate. A full pairwise alignment is then made for all the segments corresponding to the candidate SCFs. If there is a sufficiently good match among the full alignments it will be assigned to the read.
Once both V and J segments are identified, only valid matches are kept:
- the V and J segments are for the same chain;
- the J segment is located on the read after the V segment.
The D and C segments are then identified for the reads with assigned V and J segments.
For D segments, a local alignment is performed between the region of the read found between V and J, and the reference D segments for the same chain.
For C segments, the Map Reads to Reference tool is used internally. As the C segment is long and not variable, matches for the C segment for chains other than that identified for V and J indicate a false positive and the read is hence discarded.
|
The V and J segments are required for successfully clonotyping a read, because otherwise the CDR3 cannot be determined.
The D and C segments are optional. Note that the (lack of) identification of these two segment types can lead to the tool reporting clonotypes as the same or different clonotypes:
|
A read with multiple segment matches will provisionally have all these segments assigned and in a subsequent merging step, it may be assigned a specific segment.
Merging of clonotypes
After the initial identification of clonotypes, some clonotypes are merged to reduce false positives due to sequencing errors and resolve ambiguities, i.e. multiple assigned segments. Clonotype merging is performed in two steps.
The first step tries to resolve ambiguous segment assignments. Some of the reference segments have a large degree of sequence identity, e.g. in mouse a recent duplication event has resulted in multiple paralogue TCR V segments with a sequence identity of more than 97%. If a sequencing read does not cover the regions where paralogue segments differ, the segment cannot be unambiguously identified. In these cases all possible segments will be listed using a comma for separation of the different options. However, there might be reads with the same CDR3 nucleotide sequence where the segment can be uniquely determined. It is unlikely that two different clonotypes would share the same CDR3 and have almost identical segments. We thus merge a clonotype with ambiguous segments into another clonotype if it has the same CDR3 sequence and segments that are a subset of the former clonotype's segments.
The second merging step tries to correct sequencing errors in the CDR3 region, where a highly expressed clonotype would result in multiple clonotypes being reported if not corrected for. In this step, clonotypes are merged if their segments are identical and the CDR3 region is sufficiently similar. For two CDR3 regions to be deemed sufficiently similar, two types of errors are considered: errors occurring in positions of low quality and errors occurring anywhere within the CDR3 region.
Running the tool
To run Immune Repertoire Analysis, go to:
Toolbox | Biomedical Genomics Analysis (![]() ) | Immune Repertoire Analysis (
) | Immune Repertoire Analysis (![]() ) | Immune Repertoire Analysis (
) | Immune Repertoire Analysis (![]() )
)
This opens a dialog where the reads can be selected. The following options for mapping, merging and frequencies can then be configured (see figure 7.6 and fig 7.7):
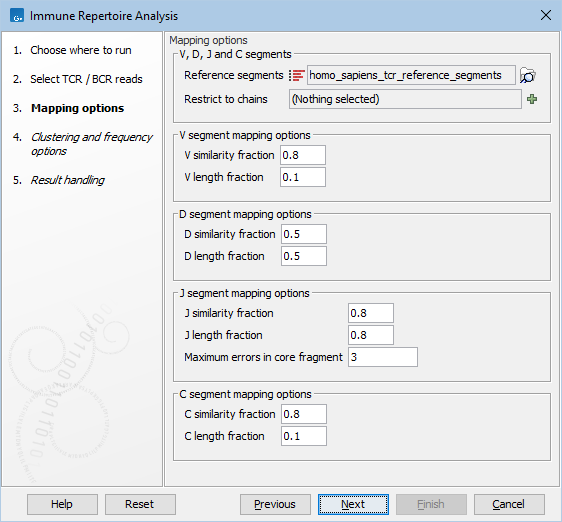
Figure 7.6: Mapping options for Immune Repertoire Analysis.
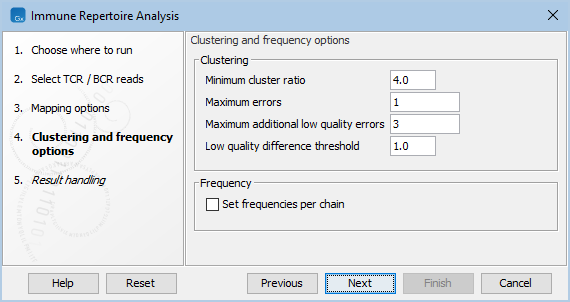
Figure 7.7: Clustering and frequency options for Immune Repertoire Analysis.
- Reference segments. A sequence list containing the V, D, J and C segments, either from the reference data or imported using Import Immune Reference Segments.
- Restrict to chains. A combination of 'TRA', 'TRB', 'TRG' and 'TRD' for TCR reference segments, or 'IGH', 'IGK' and 'IGL' for BCR reference segments, can be chosen. Only clonotypes for the selected chains will be identified. If left empty, all chains will be used.
It can be useful to set this option when only specific chains have been sequenced, to remove false positives.
- V / D / J / C similarity fraction. Minimum identity fraction between the aligned region of the read and the segment.
- V / D / J / C length fraction. Minimum fraction of the segment that must match the read.
- Maximum errors in core fragment.
Maximum number of errors allowed in the Segment Core Fragment (SCF) used for finding segment candidates for full pairwise alignment.
- Minimum cluster ratio. A smaller clonotype is merged into a larger clonotype if the number of fragments associated with the larger clonotype is at least this number of times larger than the number of fragments in the smaller clonotype.
E.g. if the minimum cluster ratio is 4 and a clonotype has 8 fragments, only clonotypes with at most 2 (8 / 4 = 2) fragments will be considered for merging. A fragment represents one single read or a pair of reads.
- Maximum errors. Two clonotypes will be considered for merging if there is at most this number of differences between their CDR3 nucleotide sequences.
- Maximum additional low quality errors. Two clonotypes where the number of differences between their CDR3 sequences exceeds ``Maximum errors'' can still be considered for merging, if the number of additional errors at positions with low quality in the smaller clonotype does not exceed this number.
- Low quality difference threshold. A position is considered of low quality if the average quality is more than this number of standard deviations lower than the average quality at each position in the CDR3 sequence.
- Set frequencies per chain. Clonotype frequencies (see Table For Clonotypes) are calculated such that they add up to 100% across all chains. If Set frequencies per chain is ticked, the frequencies are instead calculated such that they add up to 100% for each individual chain.
|
The optimal values for the Similarity fraction and Length fraction are different for the different segment types.
As the V and C segments are at the ends of the read, they might not be covered entirely and the length fraction is expected to be considerably smaller than one. On the other hand, the length fraction would typically be close to one for J. For V, J and C, the similarity fraction is usually close to one as not a lot of mutations are expected in these segments.
As the D segment is located in a region of high variability, both the similarity and length fractions would typically be lower to account for the high mutation rate. |
Subsections