Manual Annotation
This section showcases the different functionalities available from a Dimensionality Reduction Plot using a UMAP plot of an Expression Matrix (The cells in a plot can be colored using different sources of information, such as Cell Clusters, Cell Annotations, features expression and sample of origin. To enable this coloring, the relevant elements are associated with the plot by dragging and dropping them in the corresponding groups of the Side Panel (see figure 17.1).
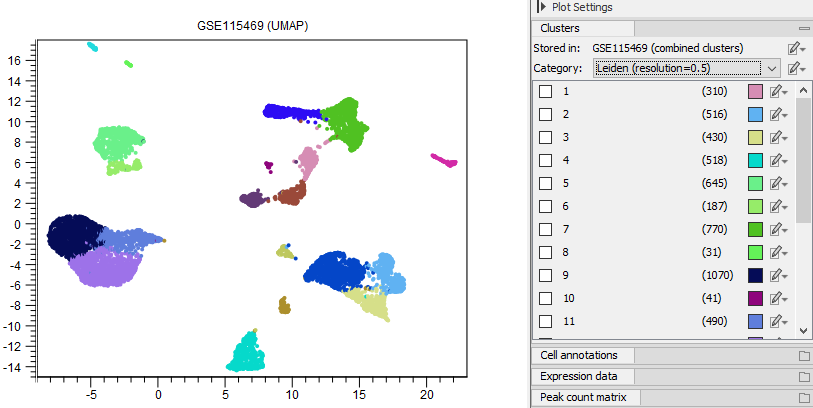
Figure 17.1: Cells are colored by clusters produced by Cluster Single Cell Data (see Cluster Single Cell Data). The category 'Leiden (resolution=0.5)' was chosen in the Side Panel Clusters group.
To visualize the same Dimensionality Reduction Plot using different sources of information for coloring, the plot can be opened multiple times and the windows can be rearranged by dragging and dropping (see figure 17.2).
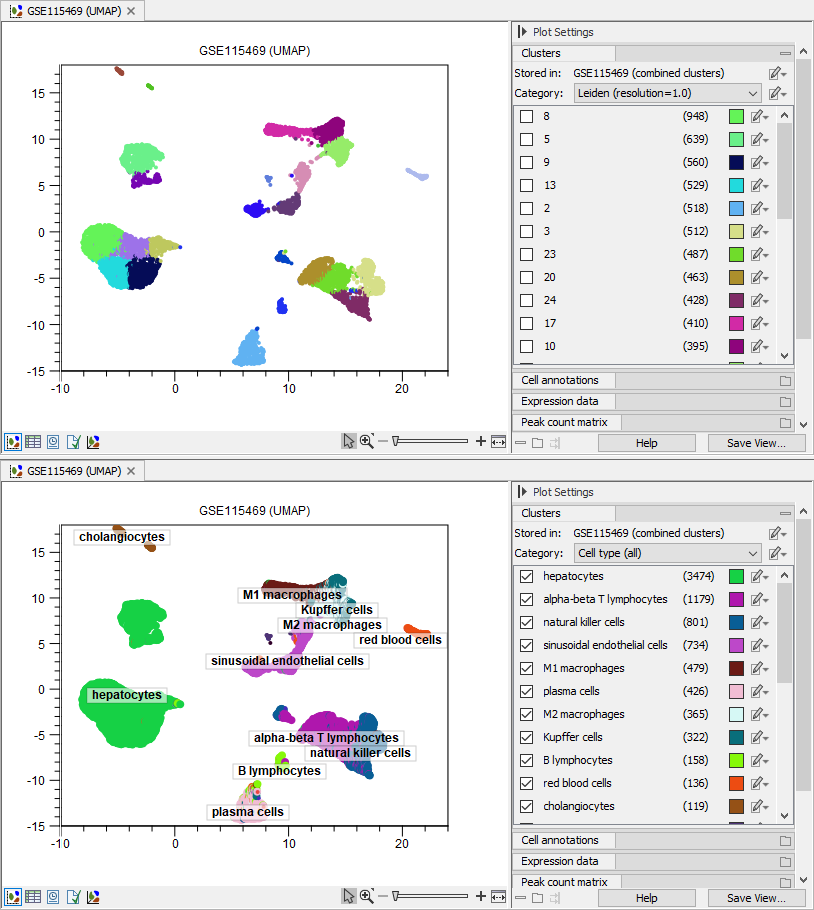
Figure 17.2: The same Dimensionality Reduction Plot is opened two times, and cells are colored by the automated clusters with 'Leiden (resolution=1.0)' (top) and the predicted cell types (bottom) produced by Predict Cell Types (see Predict Cell Type) using the human pre-trained cell classifier (see The Reference Data Manager). The label for the selected cell types is added on the plot by choosing 'Show labels from: Clusters' in the 'Coloring and highlighting' Side Panel group.
On mouse hover, a tool-tip shows summaries for the nearby cells (see figure 17.3). The same type of summary can be obtained for a group of selected cells by choosing 'Show Information for Selected' from the plot right-click menu (see figure 17.4).
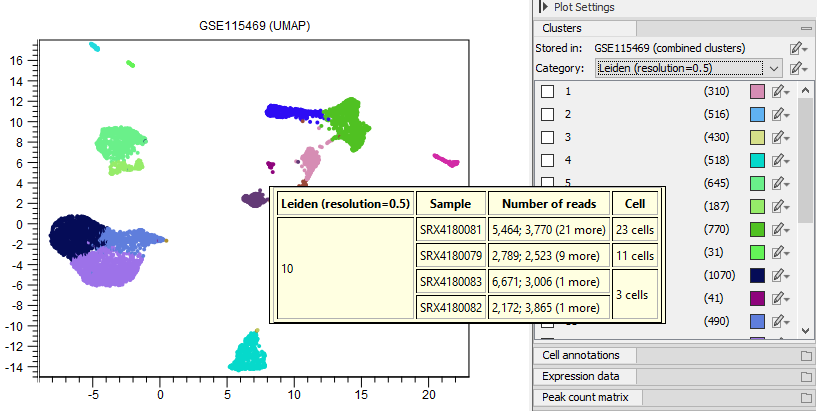
Figure 17.3: When the mouse hovers over the plot, a tool-tip is displayed summarizing the nearby cells, containing information from the elements associated to the plot, and the feature expression for any selected features.
On right-click on the plot, a series of options are available for launching tools or performing various actions on selected cells (see figure 17.4).
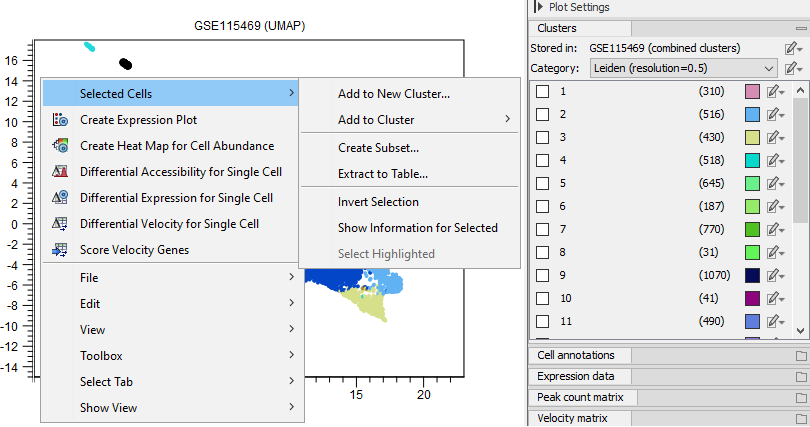
Figure 17.4: Available options in the plot right-click menu.
Selecting cells
Cells can be selected in multiple ways:
- Cells can be highlighted using the Side Panel, for example by choosing one or multiple clusters, using specific ranges for cell annotations (see figure 17.11) or feature expression (see figure 17.13), or choosing specific samples. Once the cells are highlighted, they can be selected by choosing 'Selected Highlighted' from the plot right-click menu (see figure 17.4).
- Cells can be selected using the lasso tool (see figure 17.5).
- A larger amount of cells can be selected by making a small selection and choosing 'Invert Selection' from the plot right-click menu (see figure 17.4).
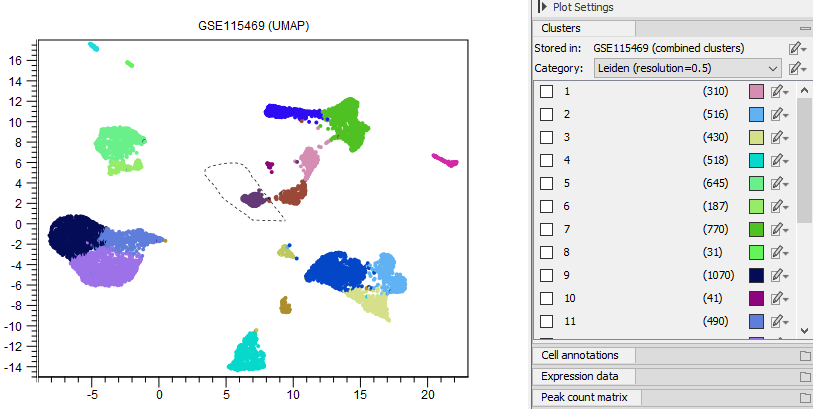
Figure 17.5: Cells can be selected using the lasso tool.
Working with clusters and annotations
Selected cells can be reassigned to existing clusters by choosing 'Add to Cluster' or added to a completely new cluster by choosing 'Add to New Cluster' (see figure 17.6) from the plot right-click menu (see figure 17.4), either as a free text or a cell type from the QIAGEN Cell Ontology (see The QIAGEN Cell Ontology). When reassigning to existing clusters, the clusters are listed in the same order as in the Side Panel (see figure 17.8).
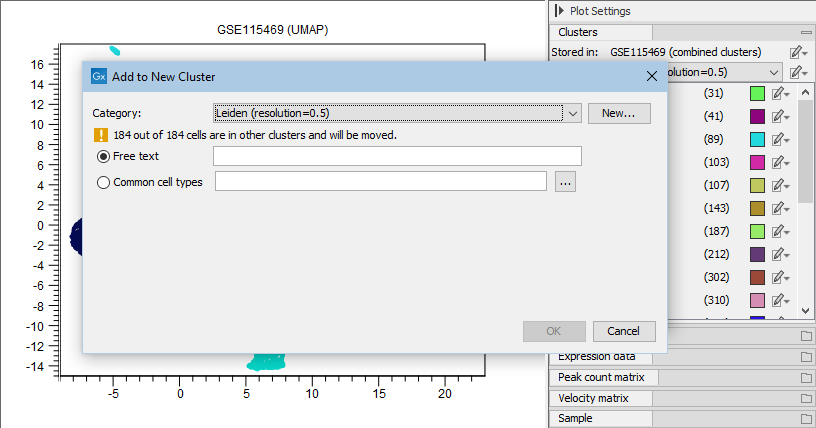
Figure 17.6: Cells can be added to a new cluster defined either by using free text or a cell type from the QIAGEN Cell Ontology by choosing 'Common cell types'. Clicking on the browse button ('...') opens up the ontology browser (see figure 17.7).
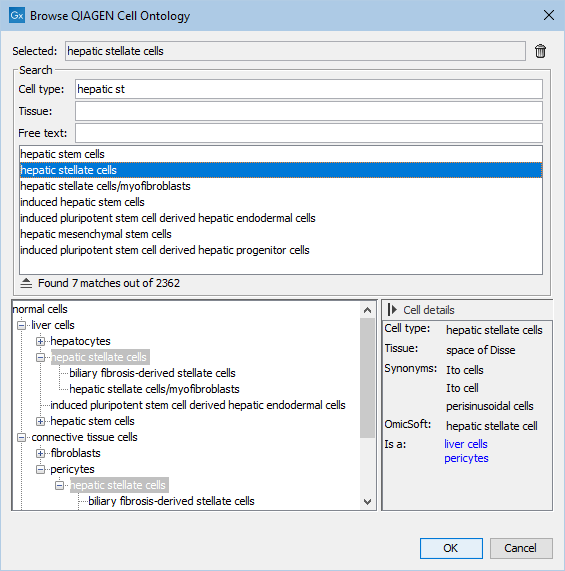
Figure 17.7: View of the QIAGEN Cell Ontology, showing details for 'hepatic stellate cells'. The 'Cell type' text field can be used to quickly identify the desired cell type. Only cell types specific to a certain tissue can be shown by filling in 'Tissue'. By using 'Free text', all cell types not containing the given text anywhere in their details are removed from the ontology structure shown at the bottom.
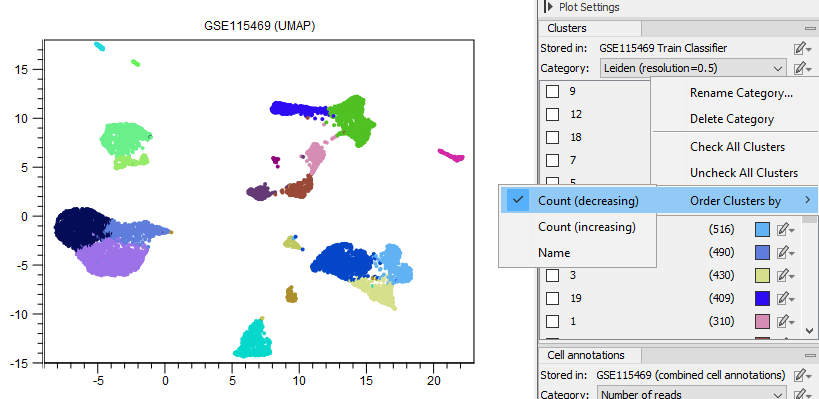
Figure 17.8: Setting the order of clusters in the Side Panel so the clusters with most cells are listed first.
An existing cluster can be renamed by choosing 'Rename Cluster' in the cluster edit menu (see figure 17.9). A dialog similar to that in figure 17.6 opens, where the cluster can be renamed either using free text or a cell type from the QIAGEN Cell Ontology. When a cluster represents an ontology cell type, details about it (as those shown in figure 17.7) can be obtained by choosing 'Show in QIAGEN Cell Ontology' (see figure 17.9).
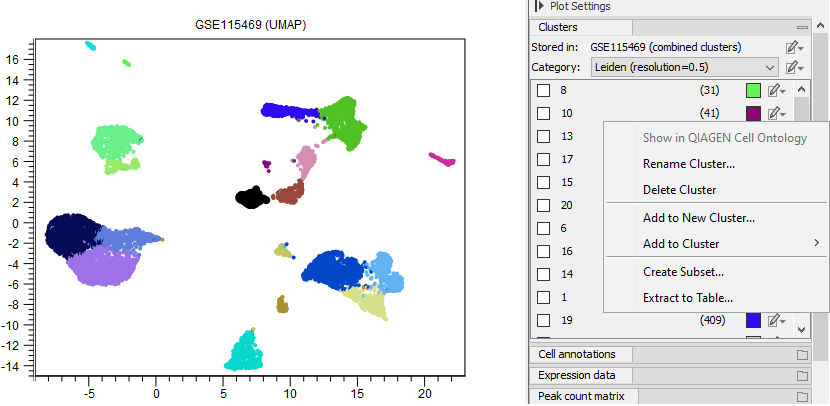
Figure 17.9: The available options for editing a single cluster from the Side Panel. 'Show in QIAGEN Cell Ontology' is grayed out because the corresponding cluster is not part of the ontology.
Any of the changes made using the above actions can be undone using the 'Undo' button. When clusters are changed, the plot name is marked with an '*' indicating that it contains an element that needs to be saved. By clicking 'Save', a new Cell Clusters element can be created.
The coloring of a cluster can be changed by clicking on the color box next to its name in the Side Panel. The newly chosen color can be saved using the View Settings, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Side_Panel_view_settings.html. Note that when the plot is closed and opened again, the default color is used, and to recover the custom color, the previously saved view settings need be re-applied to the plot.
Cells can be also colored using information from cell annotations (see figures 17.10 and 17.11).
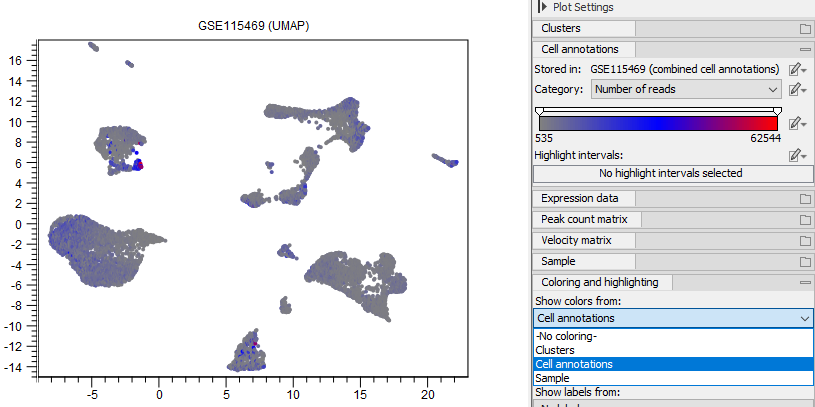
Figure 17.10: Cells are colored by the number of reads from the annotation produced by QC for Single Cell (see QC for Single Cell). What information the cells are colored by can be chosen in the group Coloring and highlighting at the bottom of the Side Panel.
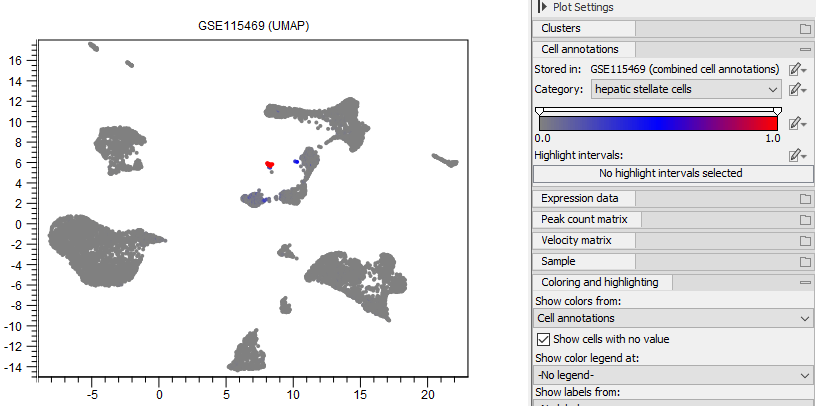
Figure 17.11: Cells are colored by the probability of having the type 'hepatic stellate cells' from the annotation produced by Predict Cell Types (see Predict Cell Type) using the human pre-trained cell classifier (see The Reference Data Manager). Cells with a probability of at least 0.3 are highlighted. The highlight interval is inclusive.
Visualizing expressions
Visualizing the expression of marker genes and selecting cells that express a set of marker genes above a specific value can be used to manually annotate the cell types. The plot can show the full expression of a specific gene across all cells using a gradient (see figure 17.12) or using one color for the cells with one or multiple gene expressions in a specific interval (see figure 17.13).
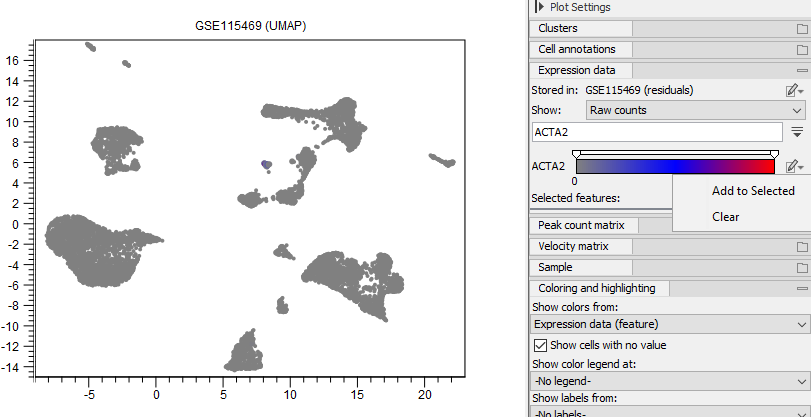
Figure 17.12: Cells are colored by the expression of 'ACTA2'. The gene is selected by typing its name in the search box under the 'Expression data' group in the Side Panel. The relative coloring of the values can be changed by dragging the two knobs on the white slider above. 'ACTA2' can be added to 'Selected features' (see figure 17.13).
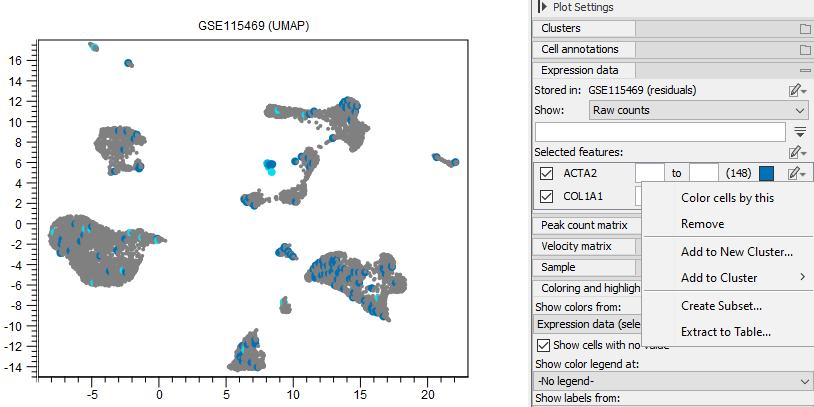
Figure 17.13: Cells that have an expression of at least 5 for 'ACTA2' and 1 for 'COL1A1' are colored in the plot. Different options are available for manipulating the cells expressing a particular feature. Selected cells can be required to express both genes, or just one of them, by choosing 'Selected in all (intersection)' or 'Selected in all (union)' at the bottom of the Side Panel. Note that the expression range is inclusive: setting the minimum expression to 0 will include the cells not expressing the gene. Choosing the interval [0, 0] will highlight only the cells that do not express the gene.
Multiple genes can be selected by:
- Adding them manually by choosing 'Add to Selected Features', see figure 17.12.
- Loading them from a file (see figure 17.14). The file should contain one gene name per line (see figure 17.15).
- Selecting them from other views showing feature expression, see below.
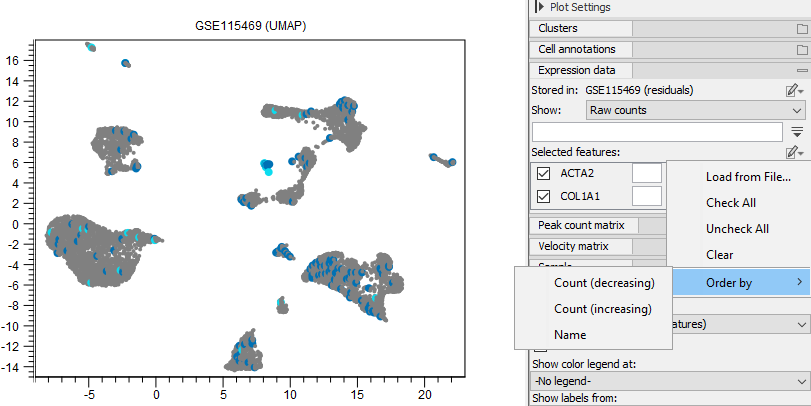
Figure 17.14: Side Panel options for 'Selected features'.
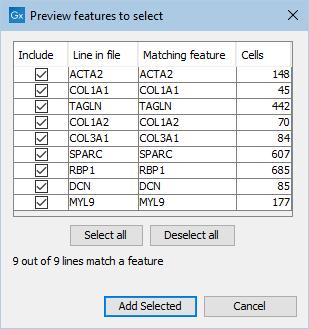
Figure 17.15: Dialog for loading feature names from a file.
Selecting features in other views
Features can be selected from various elements showing their expression, and this is done in a synchronized manner, such that all opened elements showcasing feature expression will highlight the corresponding features, if available. This can be done from an Expression Matrix (see figure 17.16), a Dot Plot (see figure 17.17), a Heat Map (see figure 17.18), or a Violin Plot (see figure 17.19).
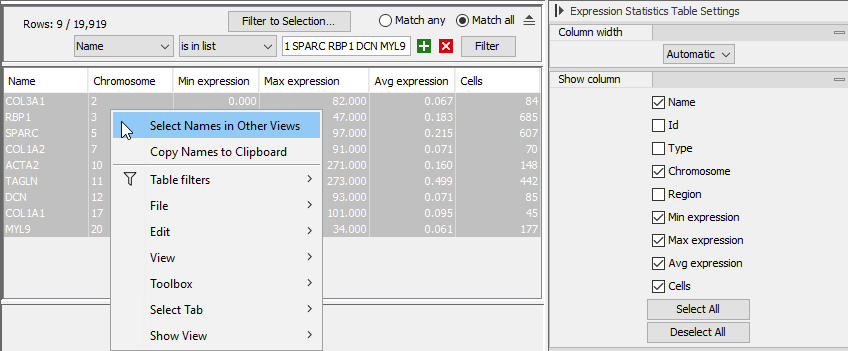
Figure 17.16: Right-click menu for selecting genes from the Expression Statistics Table view of an Expression Matrix to be synchronized with other views.
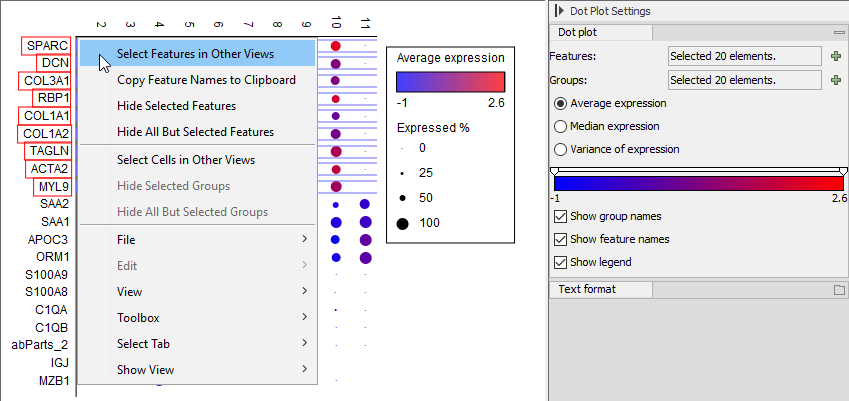
Figure 17.17: Right-click menu for selecting genes from a Dot Plot to be synchronized with other views.
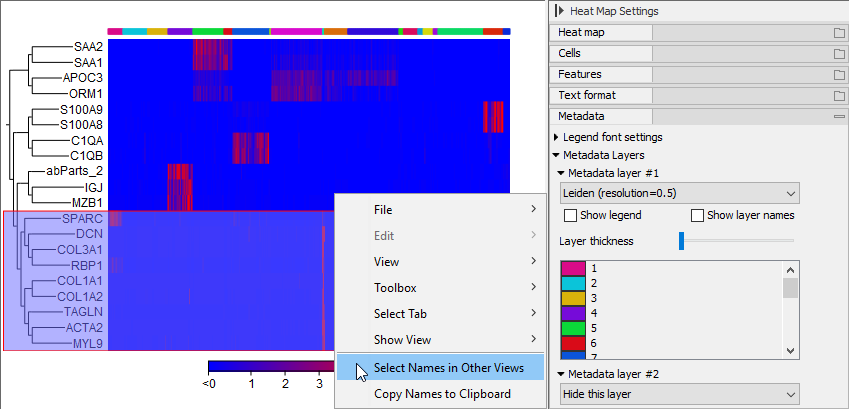
Figure 17.18: Right-click menu for selecting genes from a Heat Map to be synchronized with other views.
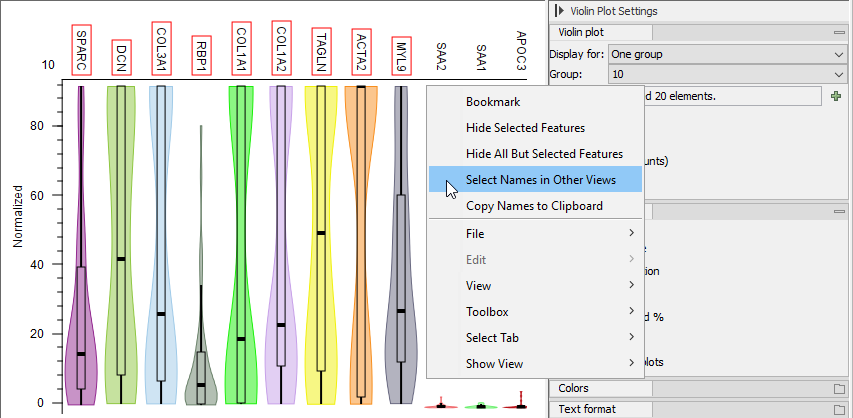
Figure 17.19: Right-click menu for selecting genes from a Violin Plot to be synchronized with other views.
The UMAP, Dot Plot, Heat Map, and Violin Plot all show that 'ACTA2', 'COL1A1', 'TAGLN', 'COL1A2', 'COL3A1', 'SPARC', 'RBP1', 'DCN', 'MYL9' are highly expressed in cluster 10. These genes were identified as markers for hepatic stellate cells [MacParland et al., 2018] and cluster 10 is confirmed to contain hepatic stellate cells by the Predict Cell Types tool (see figure 17.11). This can be further confirmed by investigating a Cell Abundance Heat Map (see Create Heat Map for Cell Abundance), as shown in figure 17.20.
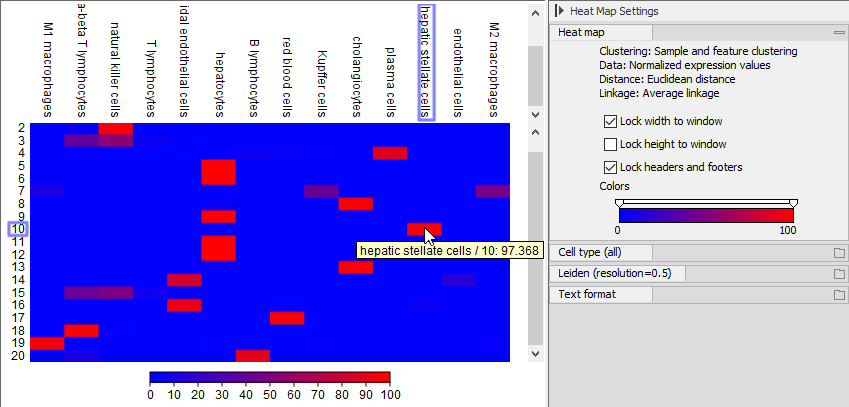
Figure 17.20: Cell Abundance Heat Map comparing the 'Leiden (resolution=0.5)' clusters to the cell types produced by Predict Cell Types. Hovering over a rectangle reveals the abundance of the selected combination.
Once the cells co-expressing specific markers are highlighted, a new cluster with the corresponding cell type can be created, as described above.