Import Peak Count Matrix
The following peak matrix formats can be imported into a Peak Count Matrix (
- Cell Ranger HDF5
- MEX
- MEX archive
Options common to all importers
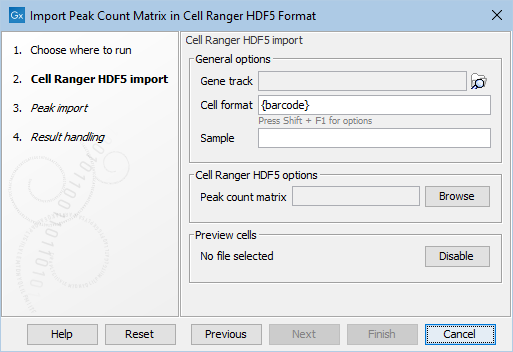
Figure 2.2: The Cell Ranger Peak Count importer. The General options are common to all the peak matrix importers.
The first step of all importers take the following options as shown in Figure 2.2:
- Gene track Positions in the imported data are matched with the provided track.
Matching is used to:
- View the Peak Count Matrix as a Track. For more information on tracks, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Tracks.html.
- Identify nearby genes if these are not explicitly supplied.
- Cell format and Sample: How cells are identified. See Cell format in importers for more details.
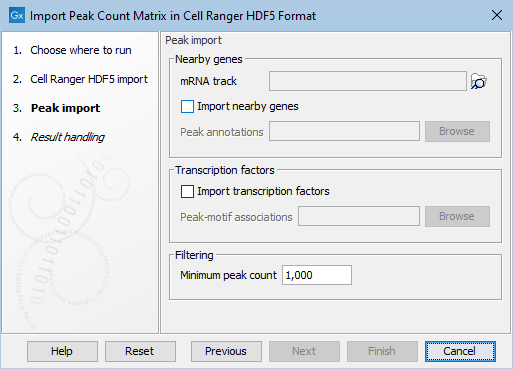
Figure 2.3: Additional options common to all the peak matrix importers.
The second step of all importers take the following options as shown in Figure 2.3:
- Nearby genes Nearby genes are determined in one of two ways:
- By searching for nearby genes using the Gene track and an accompanying mRNA track
- By supplying nearby genes in a selected tab-separated file (.tsv).
The file must consist of either:
- 6 columns: name of the chromosome prefixed with "chr" (e.g., "chr1"), start and end position of the peak, the name of the gene, distance and type of peak.
- 4 columns: name of the chromosome together with start and end positions of the peak (e.g., "chr1:123-456"), the name of the gene, distance and type of peak.
The first line must be column headers.
The distance is the number of base positions from the start or end of the peak to the start or end of the gene, whichever is closest. It is signed and will be negative if the peak is before the gene.
The type of the peak can be either "promoter" or "distal". Other values are ignored.
If there are multiple nearby genes per peak, they can either be on separate lines or be grouped on one line, with gene name, distance and peak types separated by semi-colon.
- Transcription factors If enabled, transcription factors will be imported from the selected tab-separated bed file. Each line consists of the name of the chromosome (e.g., "chr1"),
start and end positions of the peak, and the name of the transcription factor.
If not enabled, the peak matrix will not have transcription factors.
The data to be imported may either consist of peak data only or it may be a mixture of peaks and gene expressions, as is the case for 10x Multiome files. In the latter case, the gene expressions must be imported into a separate Expression Matrix (see Import Expression Matrix).
Details specific to the MEX importer
The MEX importer requires three files to be supplied:
- Barcodes file A file with the extension .tsv and tab-separated columns, with one row per barcode. It can optionally contain a header. The barcodes are read from the first column. Empty lines are ignored.
Use the Cell format option to control how the barcodes should be interpreted - for example if it also includes information about the sample.
- Feature or peak file This should be one of:
- A feature file with extension .tsv and six tab-separated columns, with one row per feature or peak. These are relevant for mixtures of peaks and expressions, e.g. 10x Multiome. The columns are: identifier (e.g., "chr1:123-456"); name (same as identifier for peaks); type, e.g. "Gene Expression" or "Peaks"; chromosome (e.g., "chr1"); start and end position of the feature or peak. The file can optionally contain a header. Empty lines are ignored.
- A peak file with extension .bed and three tab-separated columns: the chromosome, start and end position.
- Matrix file A file containing the expression with the extension .mtx in the Matrix Market Exchange Coordinate Format.
See https://math.nist.gov/MatrixMarket/formats.html for details of the Matrix Market Exchange Coordinate Format.
Details specific to the MEX archive importer
The MEX archive importer is provided for convenience. It accepts a .zip, .tar or .tar.gz archive file containing the files required by the MEX importer. In order to uniquely identify each file, these must have a specific name:
- Barcodes file must be named barcodes.tsv
- Feature or peak file must either be named features.tsv or peaks.bed
- Matrix file must be named matrix.mtx
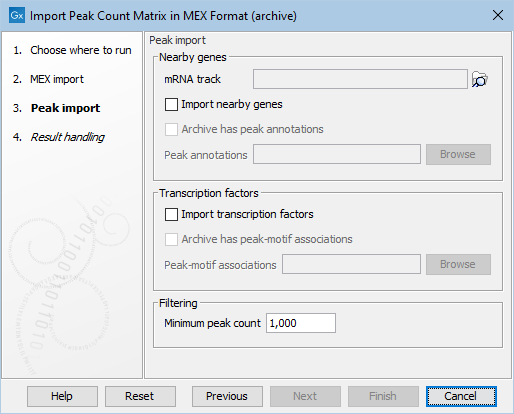
Figure 2.4: Options for nearby genes and transcription factors for the MEX archive importer
The nearby genes and/or transcription factors can be passed in as separate files as is common to all the importers.
Alternatively, they can be included in the archive file. Then that should be indicated with checkbox "Archive has peak annotations" respectively "Archive has peak-motif associations". The file names must match a specific pattern:
- Nearby genes file must end with peak_annotation.tsv
- Transcription factor file must end with peak_motif_mapping.bed
This can be relevant for importing an archive produced by the peak matrix exporter using compression (see Export Peak Count Matrix). It may also be necessary when passing multiple files to a batching or iterating workflow (see Chromatin Accessibility and Expression Analysis from Matrix).