Annotate Reads with Cell and UMI
Annotate Reads with Cell and UMI (
The tool takes as input one or more sequence lists (![]() ) of reads. For each input it outputs a list of `demultiplexed reads', which can be used in Single Cell RNA-Seq Analysis (see Single Cell RNA-Seq Analysis) and Single Cell V(D)J-Seq Analysis (see Single Cell Immune Repertoire Analysis). It optionally produces a list of reads that did not match the configured `read structure' and a report.
) of reads. For each input it outputs a list of `demultiplexed reads', which can be used in Single Cell RNA-Seq Analysis (see Single Cell RNA-Seq Analysis) and Single Cell V(D)J-Seq Analysis (see Single Cell Immune Repertoire Analysis). It optionally produces a list of reads that did not match the configured `read structure' and a report.
The tool requires the read structure to be provided (figure 4.1). Default read structures are available for selected 10x Genomics protocols, for BD Rhapsody, and for QIAseq UPX 3' protocols.
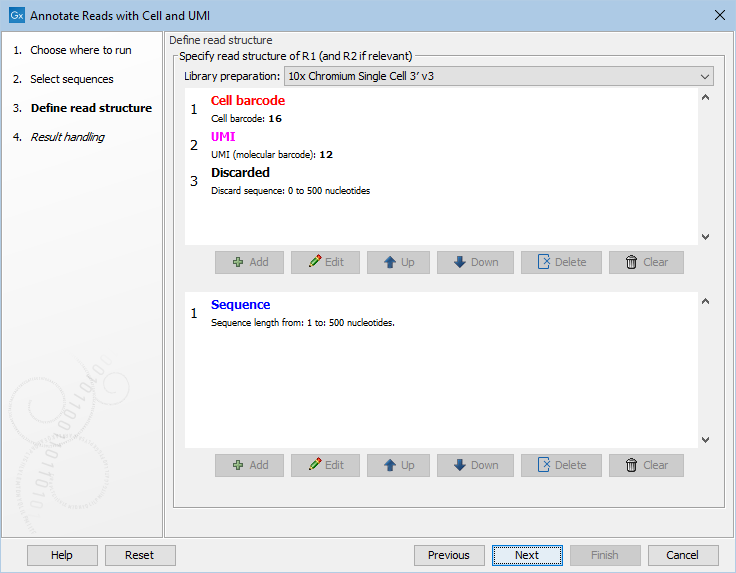
Figure 4.1: Defining the read structure for a 10x 3' gene expression protocol.
The sample name used for the output can be set using the `Sample name' option, see Setting the sample name for details.
Custom read structures
To supply a custom read structure, select Custom from the Library preparation dropdown. This enables editing of the two panels beneath the dropdown.
The top panel should be configured to describe R1 of a pair, or single-end reads. The bottom panel describes R2. For single-end reads, the configuration in the bottom panel is ignored.
The read structure can be composed of four different types of tags:
- Discarded. The corresponding sequence will be removed from the output read.
- Cell barcode. The corresponding sequence will be removed from the output read, but it will be added as an invisible annotation on the read.
- UMI. The corresponding sequence will be removed from the output read, but it will be added as an invisible annotation on the read.
- Sequence. Only this part of the read will be retained in the `Demultiplexed reads' list.
An example read structure is shown in figure 4.1. Here R1 ends with a part of variable length from 0 nt to 500 nt. This means that R1 is specified as being 16 nt of cell barcode + 12 nt of UMI and then some unknown amount of sequence. Read pairs with an R1 shorter than 16+12=28 nt will not match the read structure, and will not be present in the output. Similarly, read pairs with an R1 that is longer than 528 nt will not be present in the `Demultiplexed reads' output. Note that only R2 has a `Sequence' part. This means that the output will be single-end reads - consisting of R2 from the original pairs - but annotated invisibly with the cell barcode and UMI that were present on R1.
When configuring the read structure, be sure to describe the full length of the read. Figure 4.2 shows a similar R1 configuration as in figure 4.1. However, because the variable length part is missing, only read pairs with an R1 that is exactly 28 nt long will match the read structure: no other read pairs will be present in the `Demultiplexed reads' output.

Figure 4.2: A partially defined read structure. This will only match reads that are exactly 28 nt long, which is unlikely to be the intended behavior. Adding a variable length part as in figure 4.1 will allow matches to reads that are also longer than 28 nt.
Structures of many different libraries are listed at https://teichlab.github.io/scg_lib_structs/. For example, at the time of writing, that resource describes Microwell-seq as having an R1 structured as 6 nt cell barcode + 15 nt adapter + 6 nt cell barcode + 15 nt adapter + 6 nt cell barcode + 6 nt UMI + polyA (R2 contains the biological insert). This would be configured as shown in figure 4.3.

Figure 4.3: An example of a possible Microwell-seq R1 configuration. A single 18 nt cell barcode will be constructed from the three shorter parts.
In the Microwell-seq example, the tool would construct a single 18 nt cell barcode from the three shorter parts. More general constructions are possible. For example, if two UMI parts are defined, one on R1 and one on R2, then a single UMI will be constructed from both parts.
|
Index reads Some library preparations result in UMIs or cell barcodes being present on index reads. For example, in Smart-seq2, the cell barcodes are the sample index. As it is not possible to specify an index read in Annotate Reads with Cell and UMI, the index reads must be prepended to the corresponding read prior to analysis. |
Multiome ATAC
If read structure `10x Chromium Single Cell Multiome ATAC' is selected then 10x Multiome ATAC barcodes are translated to 10x Multiome GEX barcodes. This makes it possible to combine ATAC and GEX reads for e.g. dimensionality reduction plots. Workflow Chromatin Accessibility and Expression Analysis from Reads illustrates this (see Chromatin Accessibility and Expression Analysis from Reads). Reads with barcodes that cannot be translated are discarded.
It is not possible to customize the read structure whilst retaining the barcode translation.
Subsections