Output from the Quantify QIAseq UPX 3' workflow
The Quantify QIAseq UPX 3' workflow generates the following outputs:
- A Gene Expression track (
 ) A track showing gene expression annotations and that can be used in subsequent analyses. If you have zoomed in to nucleotide level, a tooltip will appear with information about gene name and expression values. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Expression_tracks.html.
) A track showing gene expression annotations and that can be used in subsequent analyses. If you have zoomed in to nucleotide level, a tooltip will appear with information about gene name and expression values. See http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Expression_tracks.html.
Note that the Quantify QIAseq UPX 3' workflow run on a server will also output a Transcript Expression track and a read mapping. This track should not be used for subsequent analyses because these TE tracks assume uniform coverage along transcripts, which is not the case for UPX 3' data.
- Two trimming reports - see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Trim_output.html. One is generated after trimming short polyA sequences from the middle of R1 and discarding R2. The second is issued after trimming polyA from R1.
- A UMI Group report (
 ) containing a breakdown of UMI groups with different number of reads, along with percentage of groups and reads (see Calculate Unique Molecular Index Groups).
) containing a breakdown of UMI groups with different number of reads, along with percentage of groups and reads (see Calculate Unique Molecular Index Groups).
- A UMI Group Creation report () that indicates how many reads were ignored and the reason why they were not included in a UMI read (see Create UMI Reads).
- A RNA-Seq report () - see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=RNA_Seq_report.html. You can run the tool Create Combined RNA-Seq Report to perform quality control of all RNA-Seq reports generated in one glance.
Note that the Quantify QIAseq UPX 3' workflow discards reads that map to multiple genomic locations. This can lead to genes with homologs or containing repetitive regions being reported as having low or no expression. In order to estimate expressions including multi-mapping reads, open a copy of the workflow and untick the option "Exclude ambiguously mapped reads" in the Calculate Unique Molecular Index Groups tool.
Notes about the RNA-Seq report:
- The % of mapped reads will nearly always be 100%: this is because UMI reads are made only from those reads that mapped. These UMI reads are then re-mapped.
- In the final section of the RNA-seq report called "Transcript length coverage" (see figure 6.3), there should be a red warning for "Difference between average 3' and 5' normalized counts" as this is a 3' sequencing protocol. Similarly, the plot "Coverage among normalized transcript length" should show high coverage on the right-hand side of the plot. The red and cyan lines (transcripts of length 2001-5000 nt and 5001-10000 nt) may look more erratic, because relatively few reads map to transcripts of this length.
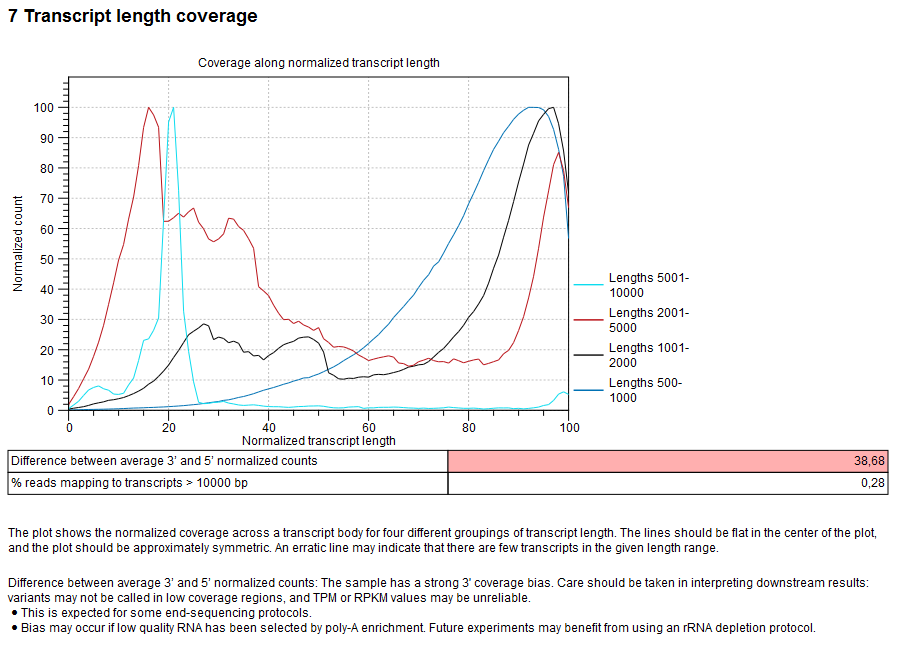
Figure 6.3: Transcript length coverage section of the RNA-seq report.
Use Gene Expression tracks to perform Differential Expression. If the analysis is set up as a single-cell experiment, a clustering of the data could provide more insights. All the tools available for downstream analyses can be found in the RNA-seq Analysis folder of the Toolbox.