Trim Primers of Mapped Single Reads
The tool Trim Primers of Mapped Single Reads removes the primer parts of reads as they reflect the primer that was added and not the actual sample.
The tool can be found in the Toolbox here:
Tools | QIAseq Panel Expert Tools | QIAseq DNA Panel Expert Tools (![]() ) | Trim Primers of Mapped Single Reads (
) | Trim Primers of Mapped Single Reads (![]() )
)
In the first dialog (figure 18.1), select a read mapping.
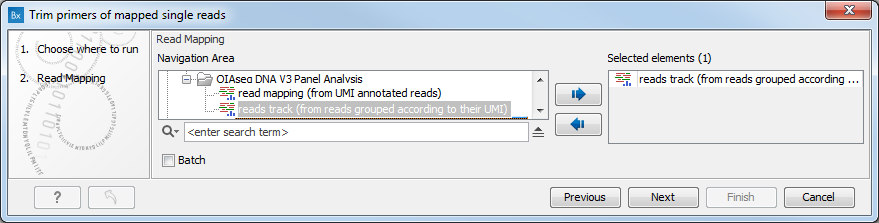
Figure 18.1: Select a read mapping.
In the second dialog (figure 18.2), select the primer annotation track that was provided with the QIAseq DNA V3 Panel.
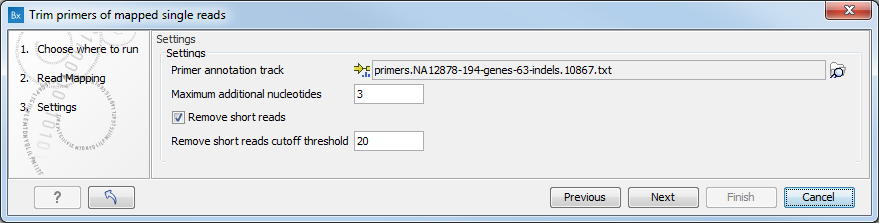
Figure 18.2: Select the primer annotation track specific to the panel.
Unlike other similar tools, this tool works on reads that potentially ends in a primer (rather than starts in a primers). The tool aims to unalign the primer parts of reads that came from that primer. It approximates this by only unaligning reads that end inside of a primer or up to 3 (set by the parameter "Extra width at end") extra bases after the primer. After unaligning the primer part of the reads, the tool removes reads where less than 20 (set by the parameter "Removal Cutoff Threshold") aligned bases remain. This feature can be disabled by unchecking the parameter "Remove Reads that are Small After unaligning".
If one read in a UMI group runs past the primer it overlaps, it means that all reads in that group were not created from that primer. If this happens, then the tool will not unalign any reads in this UMI group.