Prepare Raw Data
The first thing to do after data import is to check the quality of the sequencing reads and perform the necessary trimming. Note that the workbench is able to trim automatically read-through adapters, but if you are not sure you have read-through reads, you will need to provide a Trim Adapter List. To learn how to create an adapter trim list, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Trim_adapter_list.html.
- Go to the Toolbox and double-click on the Prepare Raw Data ready-to-use workflow (figure 12.4).
Figure 12.4: Ready-to-Use Workflows are found in the Toolbox.This will open the wizard shown in figure 12.5 where you can select the reads that you wish to prepare for further analyses.
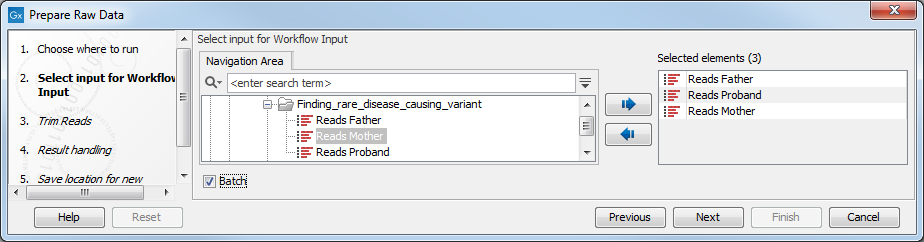
Figure 12.5: Select the sequencing raw data that you wish to prepare before further analysis. In this example we show how to prepare several reads in batch mode.There are three ways you can prepare your data: you can run them through the workflow one sample at the time, or you can select and prepare them simultaneously, or finally you can run them in batch mode. If you use batch mode, you will get an individual report for every single sample, whereas you will get one combined report for all samples if you do not run in batch mode. To run the samples in batch mode, check the "Batch" and select either several samples, or an entire folder that holds the data you wish to analyze. The next dialog recapitulates the data that will be run in batch, and let you exclude or include particular elements from the process.
- When you have selected the sample(s) you want to prepare, click Next. In the trim Reads dialog (figure 12.6) you can specify different trimming parameters and potentially select a relevant Trim adapter list.
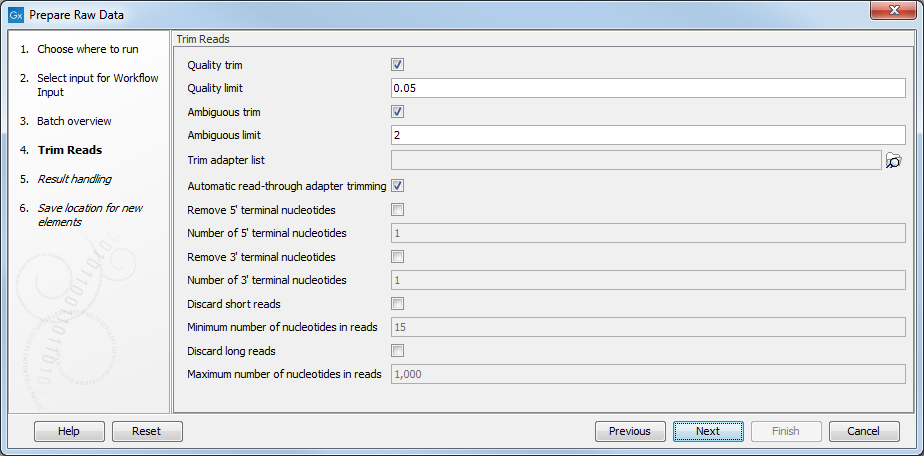
Figure 12.6: Select your trim adapter list. - In the last dialog, clicking on Preview All Parameters gives you the chance to check the selected settings. If you wish to make changes you have to use the Previous button from the wizard to edit parameters in the relevant windows. The settings can be exported with the two buttons found at the bottom of this wizard; one button allows specification of the export format, and the other button (the one labeled "Export Parameters") allows specification of the export destination. When selecting an export location, you will export the analysis parameter settings that were specified for this specific experiment.
- Click on the button labeled OK to go back to the previous wizard and choose Save the output.
Prepare Raw Data performs quality control and trimming of the sequencing reads and generates the following outputs (figure 12.7).
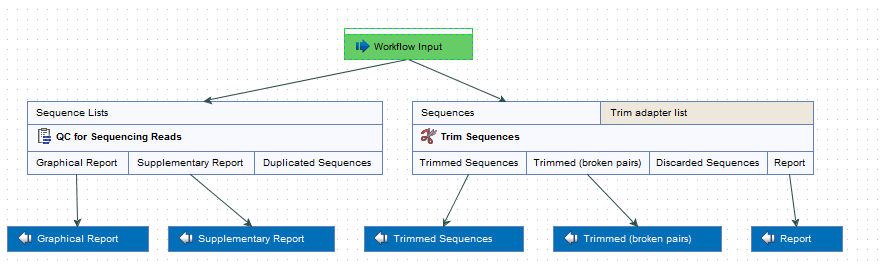
Figure 12.7: Check the settings and save your results.
- QC graphic report and QC supplementary report. For a detailed description of the QC reports and indication on how to interpret the different values, see
http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=QC_Sequencing_Reads.html.
- Trimming report. See
http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Trim_output.html.
- Trimmed sequences output. Use as input together with the "Trimmed sequences (broken pairs) output" in the next ready-to-use workflow.
- Trimmed sequences (broken pairs) output. We do not recommend to use as input in the next ready-to-use workflow.
All three reports should be inspected in order to determine whether the quality of the sequencing reads and the trimming are acceptable. The interpretation of the reports is not always completely straightforward, but as you gain experience it becomes easier. If you can accept the read quality you can now proceed to the next step and use the prepared reads output as input in the next ready-to-use workflow. If the quality of your reads is poor and cannot be accepted for further analysis, the best solution to the problem is to go back to start and resequence the sample.