Remove and Annotate with Unique Molecular Index
During library preparation of the samples with a QIAseq Targeted DNA Panel, a UMI and a common sequence prefix are added to each read before amplification. While the UMI is essential in identifying reads that originate from the same fragment, retaining it as such on the sequenced reads would hinder the subsequent read mapping efficiency and accuracy. Therefore, the Remove and Annotate with Unique Molecular Index tool removes the UMI and the common sequence prefix from the reads, while annotating each read with the UMI to retain the fragment identity as annotation.
The tool can be found in the Toolbox here:
Tools | QIAseq Panel Expert Tools | QIAseq DNA Panel Expert Tools (![]() ) | Remove and Annotate with Unique Molecular Index (
) | Remove and Annotate with Unique Molecular Index (![]() )
)
In the first dialog (figure 3.31), select the reads saved as a sequence list.
Figure 3.31: Select the reads generated using the QIAseq DNA Panel.
In the Settings dialog (figure 3.32), the following options are available.
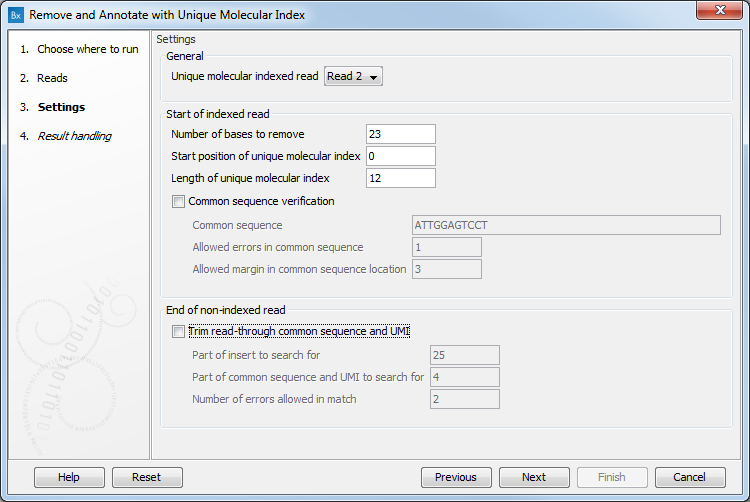
- Unique Molecular Index read defines on which read (for paired reads) the UMI will be found. For QIAseq panels, the UMI is situated on Read 2. Changing this parameter to Read 1 may compromise the results of the tools used subsequently, and in particular the results from the Calculate Unique Molecular Index Groups tool that assumes that in paired reads, the UMI will be found on Read2. Keep the value at Read 2 when using single reads.
- Number of bases to remove should be the length of the UMI and of the common sequence. It is set by default to 23.
- Start position of Unique Molecular Index is set by default to 0.
- Length of Unique Molecular Index is set by default to 12.
- Common sequence verification can be performed but not on reads generated with an Illumina Miseq sequencer. When this option is enabled, only reads for which the common sequence is recognized will be kept, and the following parameters need to be set:
- Common sequence: Type in the sequence used right next to the UMI to ensure that the UMI will be recognized as such.
- Allowed errors in common sequence: Number of insertion/deletion/mismatches we allow in the common sequence of reads and still recognize it as such.
- Allowed margin in common sequence location: Number of nucleotides that separates the actual position of the common sequence from its intended location.
- Trim read through common sequence and UMI: If this option is enabled, then for each read pair, first a sequence is extracted from the indexed read consisting of a part of the insert sequence and a part of the adjacent common sequence and UMI. Then, the reverse complement of this sequence is used to search the non-indexed read of a read pair, and if a match is found, the non-indexed read will be trimmed at the boundary between the insert and the common sequence.
- Part of insert to search for: Number of nucleotides from the sample sequence insert used to identify read-through. Increase this value to get more specific matches, decrease it if the indexed reads are very short, or to improve speed.
- Part of common sequence and UMI to search for: Number of nucleotides from the common sequence and UMI used to identify read-through. Increase this value to get more specific matches and avoid truncation at repetitive instances, decrease it to trim off shorter partial occurrences of common sequence and UMI.
- Number of errors allowed in match: Number of insertion, deletion, or mismatch errors allowed when looking for read-through sequences.
Click Next to choose whether to Open or Save your results. By default (i.e., with the Common sequence verification option unchecked), the tool outputs the same number of reads as was present in the input, where reads have had their UMI and common sequence trimmed off. It is also possible to output a report that will inform about the total number of reads processed, the total number of reads found to have UMIs and the fraction of reads that have UMIs.