Create UMI Reads for miRNA
UMI reads are created with the Create UMI Reads for miRNA tool. This tool takes a sequence list as an input (reads including UMI sequences), and outputs a new sequence list where UMI reads have been merged. In the resulting sequence list, only the small RNA sequences are present annotated with (rather than containing) the UMIs. The output of this tool can be directly used in the small RNA quantification tool.
The expected read structure of the original (untrimmed) input is illustrated in (figure 9.13). It is therefore important to trim the 5' adapter on Ion Torrent reads before running the Create UMI Reads for miRNA tool.
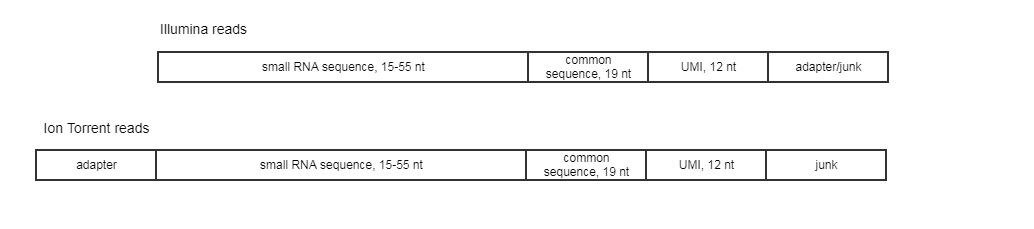
Figure 9.13: Illumina and Ion Torrent expected read structure.
The steps for merging UMI reads are as follows:
1/ The structure of the reads is analyzed. The common sequence is identified, and the small RNA sequence (preceding the common sequence) as well as the UMI (12 nucleotides following the common sequence) are identified. Reads where the common sequence is not found, or where the lengths of the small RNA or UMI do not fulfill the criteria are discarded. Note that it can be configured whether the common sequence should match exactly or whether mismatches - and how many of them, and whether these include indels - are allowed.
2/ Reads are grouped into UMI read groups based on exact identity of the small RNA sequence and the UMI. Each UMI read group keeps track of the number of reads merged into it, as well as the average nucleotide-level quality scores (if any) both for the small RNA sequence and the UMI part.
3/ We then attempt to merge "singleton" UMI read groups (containing only 1 read) into one of the existing UMI read groups based on how close the UMI and sequence match. The max number of mismatches for UMIs is set to 1. In addition, as is the case with the Create UMI Reads tool (Create UMI Reads tool):
- UMIs are matched by SNVs first, then indels if enabled.
- If there are multiple best groups to merge into, the one with the most reads will be picked. In case of a tie an arbitrary existing group is selected, unless the option "Only merge into unique UMI group matches" is checked - in which case the singleton read is not merged.
- A perfect match on UMIs with an acceptable match in the sequence supersedes always perfect match in sequence with an acceptable match on UMIs. The reason to prefer the perfect UMI over perfect sequence is that we expect the UMI is only affected by random errors (sequencing errors), whereas the miRNA sequence is affected by both random errors and the systematic/biological variations in the small RNA sequence that we actually want to be able to detect. Therefore we also expect that the "mismatch rate" will be higher in the sequence part than in the UMI part.
- We do not try to merge into groups with ambiguous bases, but we do allow them in the singleton groups to be merged. In this case they are just treated like any other SNV/indel.
Each resulting UMI read group produces one read in the output sequence list. Details on the statistics can be studies in the generated report.
To start the tool, go to:
Tools | QIAseq Panel Expert Tools | QIAseq miRNA Panel Expert Tools | Create UMI Reads for miRNA (![]() )
)
In the first dialog, choose the sequence list containing miRNA reads including UMI sequences as input. Then click Next to configure the following parameters (figure 9.14):
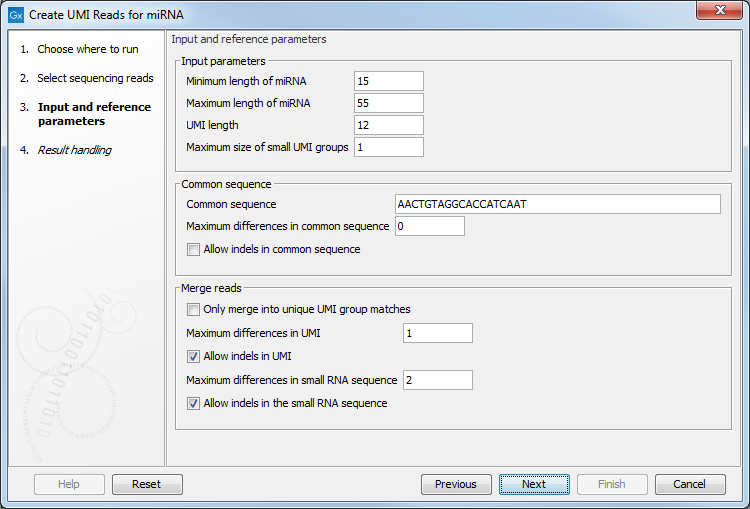
Figure 9.14: Input and reference parameters for the Create UMI Reads for miRNA tool.
- Input parameters
- Minimum length of miRNA: miRNA shorter than this value will be discarded.
- Maximum length of miRNA: miRNA longer than this value will be discarded.
- UMI length: set to 12, the size expected when using a QIAseq miRNA protocol.
- Maximum size of small UMI groups: UMI groups are split in 2 categories, small and large. Large UMI groups contain more reads than specified by this parameter, and these will not be merged. Setting this value to 1 means that the tool will only attempt to merge singletons.
- Common sequence
- Maximum differences in common sequence: maximum number of mismatches allowed in the common sequence.
- Allow indels in common sequence
- Merge reads
- Only merge into unique UMI group matches: When enabled, this option means that a small UMI group will only be merged into a large one if there is only one candidate with the best score. Unchecking this option means that a small UMI group will be merged into one of the large one even if multiple candidates are found with the same score.
- Maximum differences in UMI: Number of allowed differences in the UMI sequence when merging UMI groups. Note that this value can only be set to zero or one. As indels also count as a variation, it does not make sense to allow indels and have the number of variations be zero.
- Allow indels in UMI
- Maximum differences in small RNA sequence: Number of allowed differences in the miRNA when merging UMI groups.
- Allow indels in small RNA sequence
The tool will output a read mapping of UMI reads, i.e., a read mapping of the merged UMI groups. In the last dialog, choose whether you would like to output a report that will indicate how many reads were ignored and the reason why they were not included in a UMI read. You can also output a file containing the discarded reads before opening or saving your results.
Consensus nucleotide calculation is performed following the method described in [Hiatt et al., 2013], and can be summarized as follow:
- The UMI is defined by the majority regardless of quality. So if there are two reads with UMI ACG and one with ATG the UMI becomes ACG (there is tolerance for one SNV).
- In case of a draw, whichever read is first wins. So ACG, ATG, ACG and ATG becomes ACG whereas shuffling the order to ATG, ACG, ATG and ACG gives a consensus ATG.
- The quality is taken from the max quality of a read with exactly that UMI, so ACG (q 20), ATG (q 40), ACG (q 30) and ATG (q 50) becomes ACG (q 30). On the other hand shuffling the order to ATG (q 40), ACG (q 20), ATG (q 50) and ACT (q 20) makes it ATG (q 50). Variations have no negative impact on quality, so in the two cases above the q-score would be the same on all three nucleotides.