How to create a custom panel analysis workflow
For most applications (DNA, RNAscan, RNA, Targeted TMB/MSI, and Targeted Methyl) it is possible to set up a custom workflow.
Click on the Add custom panel button to configure the new panel (figure 2.10).
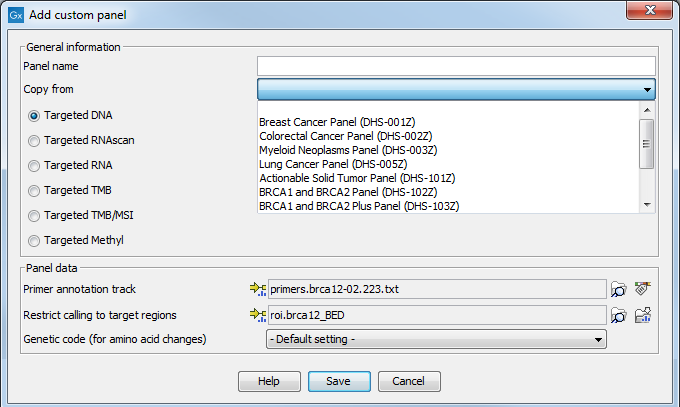
Figure 2.10: Configure the workflow with the files corresponding to your custom panel. Here we can see an example where the drop down menu "Copy from" shows all the possible custom Targeted DNA workflows available.
In the General information section, enter the name of the custom panel. Choose the application for the panel. It is also possible to use a pre-existing panel as a template by selecting it from the drop down menu. In that case, the name of the panel chose in the drop-down menu will fill the "Panel name" field, but can also be re-edited for clarity purposes. In the case of Targeted DNA applications, copying an existing panel is the easiest way to set up a workflow in which you have control over the filtering steps.
In the Panel data section, specify the following files (the type of files you have to provide depends on the application):
- Primer annotation track (Targeted DNA and RNAscan) The QIAseq DNA Panels primers are made available to you upon purchase of a kit. The Import icon allows you to import in the workbench a QIAGEN primers file previously saved on your computer (see Import QIAGEN Primers), while the Browse icon allows you to specify a file already stored in your Navigation Area.
- Restrict calling to target regions (Targeted DNA and Targeted RNA) The QIAseq DNA Panels primers target different regions of the genome that are specified in a *.bed file. This file was made available to you upon purchase of a kit. The Import icon allows you to import in the workbench a target regions BED file previously saved on your computer, while The Browse icon allows you to specify a file already stored in your Navigation Area. Note that when importing, you also have to specify also a reference sequence (hg19 for targeted DNA panels, hg38 for Targeted RNA panels). This reference sequence is pre-set once you have downloaded and applied a Reference Data Set as described in Data management.
- Genetic code (for amino acid changes) (Targeted DNA, TMB and TMB/MSI) Choose the correct genetic code, i.e., standard in most case, and Vertebrate Mitochondria when working with Mitochondria panels (DHS-105Z for example).
- Known Fusions (Targeted RNAscan only) This track will be used to annotate the fusion track output by the workflow. The Import icon allows you to import in the workbench a Known Fusion Information track (see Import Known Fusion Information Track). The Browse icon allows you to specify a file previously saved in the Navigation Area.
- Human or Mouse (Targeted RNA only): Specify the species of the samples that will be analyzed with your custom workflow.
- Masking regions This track describes "uncertain regions". The workflow will only keep variants that lie outside the masking regions.
- Mispriming events This track contains a predicted off-target priming locations of the original primers.
- Gene-pseudogene track This track lists gene-pseudogene links to remove reads that map well to pseudogene locations
- MSI baseline This track is generated with the Generate MSI Baseline tool, and using loci that are identified to perform consistently well during benchmarking.
Targeted DNA custom workflows include filter steps allowing the initial pool of variants found in the data (output in the Unfiltered Variants track) to be reduced to a smaller set of "reliable" variants (output in the Variants passing filters track). The filters are designed especially for the read type and application and are based on the following parameters:
- Minimum variant quality (QUAL) Measure of the significance of a variant, i.e., a quantification of the evidence (read count) supporting the variant, relative to the coverage and what could be expected to be seen by chance, given the error rates in the data. The mathematical derivation depends on the set of probabilities of generating the nucleotide pattern observed at the variant site (1) by sequencing errors alone and (2) under the different allele models of the variant caller allows. Qual is calculated as -10log10(1-p), p being the probability that a particular variant exists in the sample. Qual is capped at 200 for p=1, with 200: highly significant, 0: insignificant. In rare cases, the Qual value cannot be calculated for specific variant and as a result the Qual field will be empty. This value is necessary for certain downstream analyses of the data after export in vcf format. A QUAL value of 10 indicates a 1 in 10 chance that the called variant is an error, while a QUAL of 100 indicates a 1 in
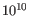 chance that the called variant is an error.
chance that the called variant is an error.
- Minimum average base quality The Avg Q of reads calculates the amount of sequences that feature individual PHRED-scores in 64 bins from 0 to 63. The quality score of a sequence is calculated as arithmetic mean of its base qualities. PHRED-scores of 30 and above are considered high quality.
- Minimum frequency (%) The frequency is calculated as
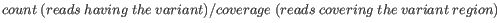
(2.1)
Only variants that are present at least at the specified frequency are called. - Minimum read direction test probability Tests whether the distribution among forward and reverse reads of the variant carrying reads is different from that of all the reads covering the variant position. This value reflects a balanced presence of the variant in forward and reverse reads (1: well-balanced, 0: un-balanced).
- Minimum read position test probability (%) The test probability for the test of whether the distribution of the read positions variant in the variant carrying reads is different from that of all the reads covering the variant position.
- BaseQRankSum Evaluation of the quality scores in the reads that have a called variant, compared with the quality scores of the reference allele. Variants for which there are no reads holding the corresponding reference allele do not have a BaseQRankSum value. The score is a z-score derived using the Mann-Whitney U test, so a value of -2.0 indicates that the observed qualities for the variant are two standard deviations below what would be expected if they were drawn from the same distribution as the reference allele qualities. A negative BaseQRankSum indicates a variant with lower quality than the reference variant, and a positive z-score indicates higher quality than the reference.
The filters remove variants that are either:
- not significant enough: filtering is performed based on the QUAL value, value that weighs count against coverage and error rate.
- likely due to artifacts: a called variant must be of sufficient quality, and have an un-biased read direction or read position presence (using the values "Average quality", "Read position test probability" and "Read direction test probability").
- homopolymer errors (indels occuring in a homopolymer stretch) using a too low frequency cut-off value.
- too infrequent.
Once your custom panel is configured, click Save to add the custom workflow to the list of workflows available from the Analyze QIAseq Panels guide. Once in the list, a button Edit allows you to change the configuration of the custom workflow. Click Run to follow the instructions given in each dialog (see Start a QIAseq panel analysis).
Subsections