Output from the Detect QIAseq RNAscan Fusions workflow
The Detect QIAseq RNAscan Fusions workflow's main outputs are two Genome Browser Views.
Genome Browser View (WT) displays the following tracks:
- Reference mRNA tracks
- Reference gene tracks
- Reference sequence tracks
- The mapping of the UMI reads
- The detected fusion genes track (based on the wild type genome sequence as reference))
Genome Browser View (Fusions) displays the following tracks:
- The detected fusion genes track (based on a fusion genome sequence as reference)
- The mapping of the UMI reads
- Fusion sequence tracks
- Fusion mRNA tracks (with a representation showing no intron between fusion exons as inserting an artificial intronic sequence could be misleading).
- Fusion gene tracks
- Fusion CDS tracks: each fusion CDS includes a "Frame aligns" annotation, which shows whether the CDS stays in the reading frame across the fusion breakpoint, allowing users to visualize fusions that are more likely to be translated to protein.
- The Fusion panel primers track
In addition, the workflow outputs a series of folders: Tracks", "Read Mappings" and "Fusions References" contains the tracks included in the Genome Browser both for Wild Type and Fusions; "QC and Reports" contain the following reports:
- A QC for Sequencing Reads Graphical Report (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=QC_Sequencing_Reads.html)
- A Remove and Annotate with UMI Report
- A Trim Adapters Report (see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Trim_output.html)
- A QC for RNAscan Panels Report, containing the following information:
- Total number of mapped reads, counting paired reads as two.
- Total number of mapped reads(-pair)s, counting paired reads as one.
- Target genes: genes targeted by the primers.
- Number of target genes.
- Primers in target genes: how many primers are within the target genes regions.
- Mean read coverage per target gene primer (exact start position match): coverage from reads that start exactly at the primer start site.
- Fraction of mapped read(-pair)s exactly matching primers [%].
- Primers outside gene regions: gDNA control primers outside target gene regions, used for detection of DNA contamination of samples.
- Mean read coverage per non-gene primer: a number higher than 0 indicates DNA contamination of the sample. A mean gDNA coverage around 50 reads or higher may increase false positive signal level.
- DNA contamination [%]: calculated using the Mean read coverage per target gene primer and the Mean read coverage per non-gene primer metrics. In general, if the Mean read coverage per non-gene primer metric is around 50 or higher, the chances for a false positive may be higher.
- Primers in reference genes [COPA, MRPS14, CIAO1, UBE3C]: QIAseq RNAscan panels typically include four reference gene primers.
- Mean read coverage per reference gene control primer: the reference genes should have a mean coverage of at least 300 reads, otherwise the effective input is too low, and false negatives are expected.
- Target gene versus reference gene coverage ratio [%]
Note about interpreting the QC for RNAscan Panels Report The relationship between input, amplification and therefore number of reads per UMI with the original coverage is complex. For example, a "good" original coverage can be deceiving, if the user has amplified many times the original molecule: in that case, the information added to the experiment is close to null.
The easiest way to review the results is to open the Genome Browser Views.
The read mapping track displays the mapping of UMI reads to the transcript sequences. It contains only reads that provide conclusive evidence for or against fusion (figure 4.4).
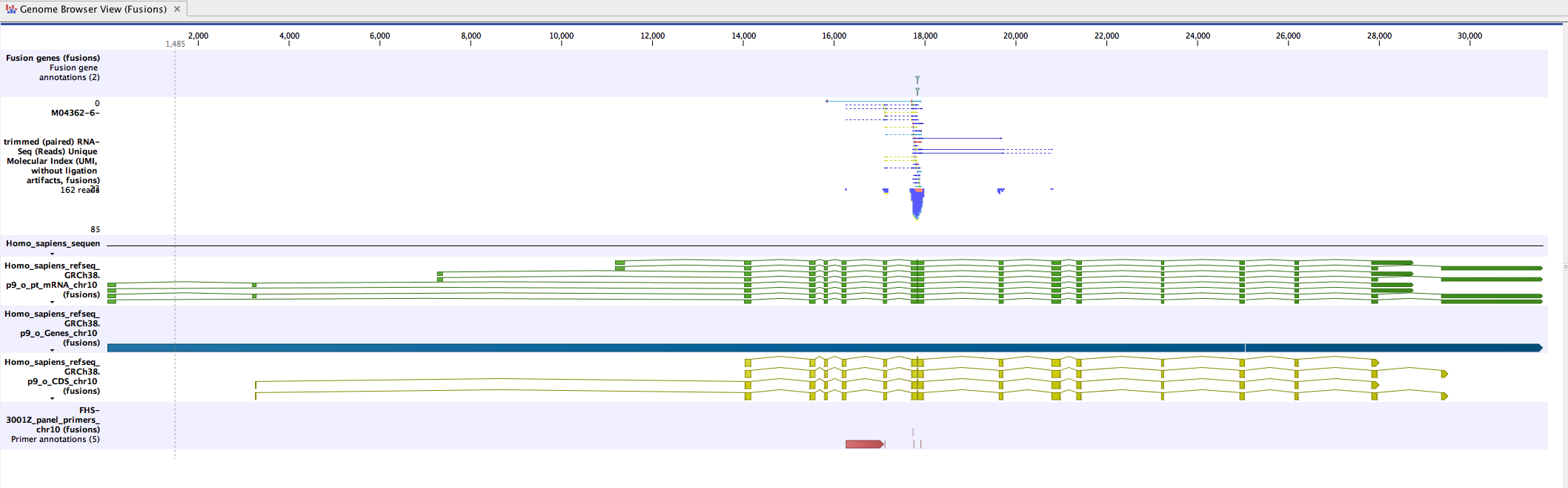
Figure 4.4: An example of fusion in a Genome Browser View.
Double-click on the fusion track name (to the right of the Genome Browser View) while pressing the Ctrl button. The fusion track will open as a table in split view, below the Genome Browser View. Clicking on a fusion event in the table will zoom in to its location in the UMI read mapping, allowing you to review the UMI reads supporting the detected fusion. Note that each fusion event is represented by two lines in the table sharing the same Fusion number. Each line corresponds to a fusion breakpoint.
Note about exporting output files in SAM/BAM format Exporting UMI reads as BAM files will show UMI described as Unique_Molecular_Index=[number1]_count=[number2], where number1 is a UMI ID (just a unique UMI group number), and number2 is the number of reads that are in that UMI group.
The fusion table contains the following information:
- Chromosome. Chromosome where "Gene" and "Transcript" are located.
- Region. Breakpoint position of the fusion event relative to the reference sequence hg38.
- Name. Short name of the fusion event, 5' gene-3' gene.
- Fusion number. This number allows to identify easily the two rows that represents a single fusion event.
- 5' or 3' Gene. The fusion gene that corresponds to the "Chromosome" and "Region" fields.
- Breakpoint type. 3' or 5'.
- Fusion Crossing Reads. Number of reads crossing the fusion breakpoint.
- 5' or 3' Read Coverage. Number of reads (unaligned ends and pairs) that cover the 5' or 3'-transcript breakpoint, including normal transcripts and fusion transcripts.
- Z-score. Converted from the P-value using the inverse distribution function for a standard Gaussian distribution.
- P-value. A measure of certainty of the call calculated using a binomial test, it is calculated as the probability that an observation indicating a fusion event occurs by chance when there is no fusion. The closer the value is to 0, the more certain the call. Although one should avoid strictly interpret the p-value as the true false positive rate, our test data show that the p-value seems to be appropriately calibrated using standard parameter settings.
- Filter. Contains the names of the filters applicable to the fusion, or the value "PASS" if it passed all filters.
- Exon skipping. Whether the fusion is a same-gene fusions where the 5' breakpoint is upstream of the 3' breakpoint.
- Compatible Transcripts. All possible and known transcripts (as identified by their transcript IDs) that the fusion reads are compatible with, such as the 5' and 3' gene transcripts that include the exons which are expressed in the fusion.
- Translocation Name. Description of the fusion in the HGVS format (http://cancer.sanger.ac.uk/cosmic/help/fusion/summary) using the preferred transcript.
- Original chromosome. (only in tracks based on a fusion reference)
- Original breakpoint region. (only in tracks based on a fusion reference)
- Known Fusion. Indicates the Fusion ID Number of the matching fusion in the known fusion database. If the fusion is not found in the database, then -1 is reported. By default, the tool uses a QIAGEN known fusion database, but you can replace this database with another known fusions database relevant to your assay. Note that this database is not used to detect the fusions, but only for annotating the identified fusions output from the Detect Fusion Genes before refinement.
- Found in-frame CDS. This column is present when a CDS track was specified as input. It contains "Yes" if at least one fusion CDS that stays in frame across the fusion breakpoints has been found. Note that the in-frame calculation only takes into account the frame of the last included exon in the 5' gene and the first included exon in the 3' gene, and ignores more complex factors that might affect frame, such as frameshift mutations or stop codons due to variants around the fusion breakpoints.
- Promiscuity. Number of different potential fusion partners found for this gene.