Refine Fusion Genes
The Refine Fusion Genes tool takes as input the fusions identified by the Detect Fusion Genes tool, and re-counts the number of fusion crossing reads as well as the wildtype supporting reads using the RNA-Seq mapping against the wild type and fusion references. The fusion reference is an artificial reference sequence that "assumes" the detected fusions by generating new chromosomes corresponding to each fusion in addition to the original chromosomes (figure 4.11).
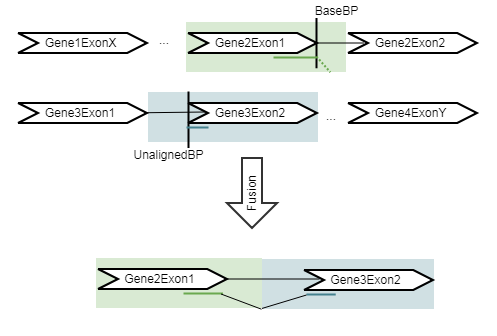
Figure 4.11: An artificial chromosome is created consisting of the vicinity of both ends of the fusion. It will be output without any intron sequence in the fusion track.
RNASeq analysis is used to re-map the reads to the artificial reference, with the expectation that reads that were used to detect the fusion will now map to the fusion transcript with a spliced read. In addition, some reads that did not originally map at all will now map to the artifical reference sequence, increasing evidence for the fusion event. The tool then calculates the "refined" Z-score and p-value using the same binomial model as the Detect Fusion Genes tool.
The Refine Fusion Genes tool can be found in the Toolbox here:
Tools | QIAseq Panel Expert Tools | QIAseq RNAscan Panel Expert Tools | Refine Fusion Genes
The Refine Fusion Genes tool takes a fusion track as input (figure 4.12).
Figure 4.12: Select a fusion track.
The next dialog of the tool allows you to configure the following parameters (figure 4.13):
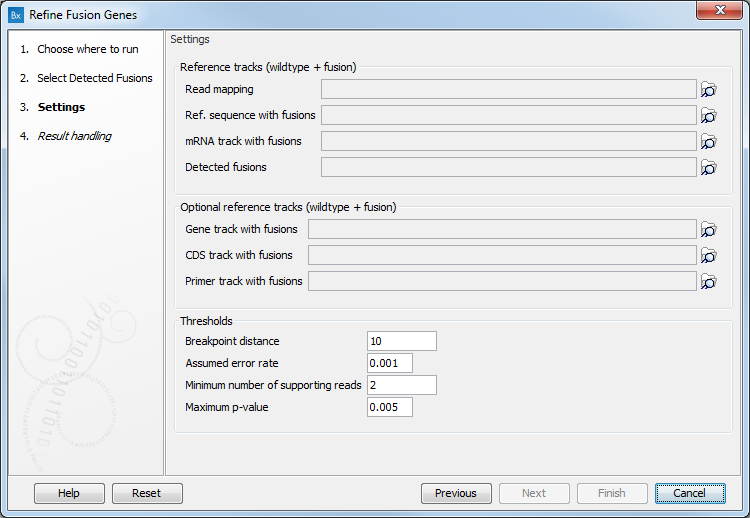
Figure 4.13: Set the parameters for the Refine Fusion Genes tool.
- Reference tracks (wildtype + fusion): Specify a read mapping, a reference sequence with fusions, a mRNA track with fusions, or a detected fusions track: these files are output by the Define Fusion Genes.
- Optional reference tracks (wildtype + fusion): You can also add a Gene track, CDS track or Primer track with fusions
- Thresholds:
- Breakpoint distance: Minimum distance from a read mapping exon position to a fusion breakpoint position required to count a read as supporting.
- Assumed error rate: Probability of an unaligned end mapping to another gene by random.
- Minimum number of supporting reads that should support a fusion. Fusions with fewer supporting reads will get a corresponding filter annotation.
- Maximum p-value: Fusions with p-value above this threshold will get a corresponding filter annotation.
The output of the Refine Fusion Genes tool is the same fusion track as the one that was input, but now including a FILTER column, where fusions "PASS" only if they fulfill the p-value and minimum number of fusion crossing reads requirements supplied by the user as parameters.