- Run the Filter Causal Variants (WES-HD) workflow
- Output from the Filter Causal Variants (WES-HD) workflow
Filter Causal Variants (WES-HD)
If you are analyzing a list of variants, you can use the Filter Causal Variants (WES-HD) ready-to-use workflow to remove variants that are outside the target region, as well as common variants present in publicly available databases. The workflow will annotate the remaining variants with gene names, conservation scores, and information from relevant databases.
Run the Filter Causal Variants (WES-HD) workflow
To run the Filter Causal Variants (WES-HD) workflow, go to:
Toolbox | Ready-to-Use Workflows | Whole Exome Sequencing (![]() ) | Hereditary Disease (
) | Hereditary Disease (![]() ) | Filter Causal Variants (WES -HD) (
) | Filter Causal Variants (WES -HD) (![]() )
)
- Double-click on the workflow name to start the analysis. If you are connected to a server, you will first be asked where you would like to run the analysis.
- Select the variant track you want to use for filtering causal variants (figure 14.44).
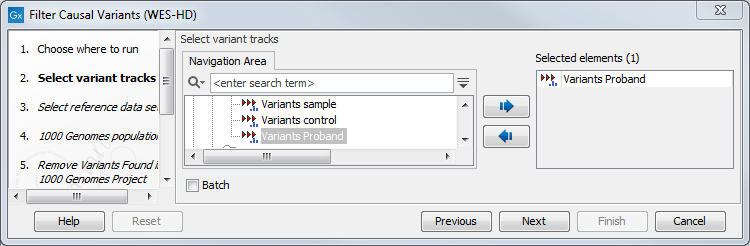
Figure 14.44: Select the variant track from which you would like to filter somatic variants. - In the next dialog, you have to select which data set should be used to filter causal variants (figure 14.45).
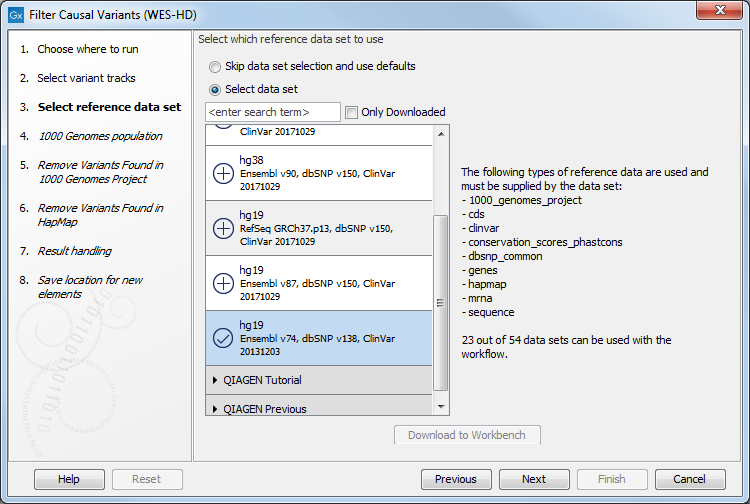
Figure 14.45: Choose the relevant reference Data Set to annotate. - Specify which of the 1000 Genomes populations should be used for annotation (figure 14.46).
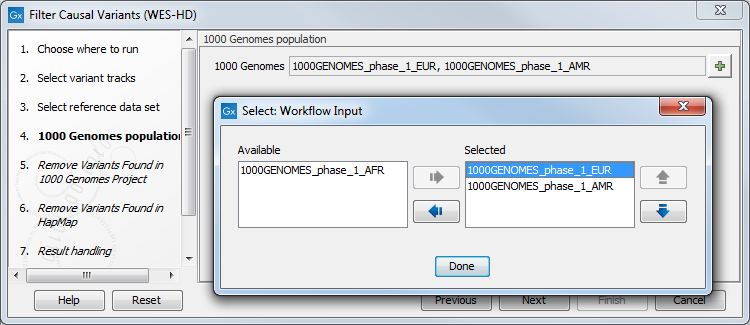
Figure 14.46: Select the relevant 1000 Genomes population(s). - In the next dialog, specify the 1000 Genomes population that should be used for filtering out variants found in the 1000 Genomes project.
- Specify the Hapmap populations that should be used for filtering out variants found in Hapmap (figure 14.47).
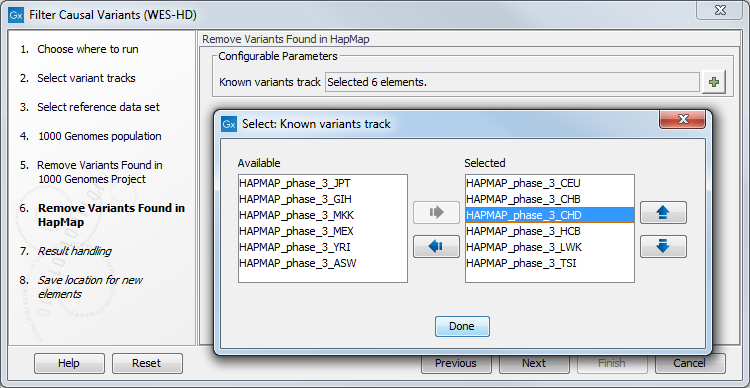
Figure 14.47: Select the relevant Hapmap population(s). - In the last wizard step you can check the selected settings by clicking on the button labeled Preview All Parameters.
In the Preview All Parameters wizard you can only check the settings, and if you wish to make changes you have to use the Previous button from the wizard to edit parameters in the relevant windows.
- Choose to Save your results and click on the button labeled Finish.
Output from the Filter Causal Variants (WES-HD) workflow
The following outputs are generated:
- An Amino Acid Track Shows the consequences of the variants at the amino acid level in the context of the original amino acid sequence. A variant introducing a stop mutation is illustrated with a red amino acid.
- A Track List
- A Filtered Variant Track