Bioinformatics explained: Multiple alignments
Multiple alignments are at the core of bioinformatical analysis. Often the first step in a chain of bioinformatical analyses is to construct a multiple alignment of a number of homologs DNA or protein sequences. However, despite their frequent use, the development of multiple alignment algorithms remains one of the algorithmically most challenging areas in bioinformatical research.
Constructing a multiple alignment corresponds to developing a hypothesis of how a number of sequences have evolved through the processes of character substitution, insertion and deletion. The input to multiple alignment algorithms is a number of homologous sequences, i.e., sequences that share a common ancestor and most often also share molecular function. The generated alignment is a table (see figure 14.19) where each row corresponds to an input sequence and each column corresponds to a position in the alignment. An individual column in this table represents residues that have all diverged from a common ancestral residue. Gaps in the table (commonly represented by a '-') represent positions where residues have been inserted or deleted and thus do not have ancestral counterparts in all sequences.
Use of multiple alignments
Once a multiple alignment is constructed it can form the basis for a number of analyses:
- The phylogenetic relationship of the sequences can be investigated by tree-building methods based on the alignment.
- Annotation of functional domains, which may only be known for a subset of the sequences, can be transferred to aligned positions in other un-annotated sequences.
- Conserved regions in the alignment can be found which are prime candidates for holding functionally important sites.
- Comparative bioinformatical analysis can be performed to identify functionally important regions.
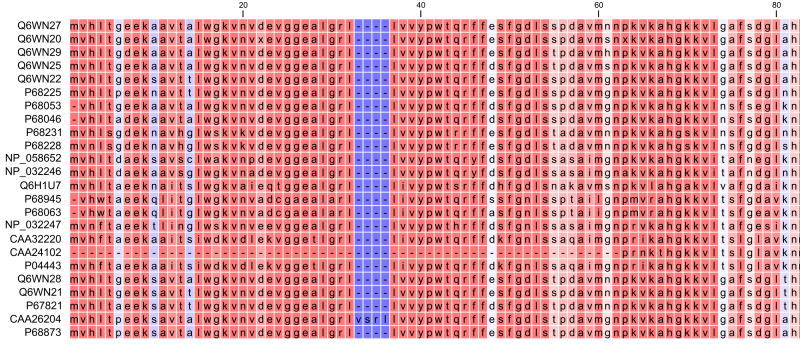
Figure 14.19: The tabular format of a multiple alignment of 24 Hemoglobin protein sequences. Sequence names appear at the beginning of each row and the residue position is indicated by the numbers at the top of the alignment columns. The level of sequence conservation is shown on a color scale with blue residues being the least conserved and red residues being the most conserved.
Constructing multiple alignments
Whereas the optimal solution to the pairwise alignment problem can be found in reasonable time, the problem of constructing a multiple alignment is much harder.
The first major challenge in the multiple alignment procedure is how to rank different alignments, i.e., which scoring function to use. Since the sequences have a shared history they are correlated through their phylogeny and the scoring function should ideally take this into account. Doing so is, however, not straightforward as it increases the number of model parameters considerably. It is therefore commonplace to either ignore this complication and assume sequences to be unrelated, or to use heuristic corrections for shared ancestry.
The second challenge is to find the optimal alignment given a scoring function. For pairs of sequences this can be done by dynamic programming algorithms, but for more than three sequences this approach demands too much computer time and memory to be feasible.
A commonly used approach is therefore to do progressive alignment [Feng and Doolittle, 1987] where multiple alignments are built through the successive construction of pairwise alignments. These algorithms provide a good compromise between time spent and the quality of the resulting alignment
The method has the inherent drawback that once two sequences are aligned, there is no way of changing their relative alignment based on the information that additional sequences may contribute later in the process. It is therefore important to make the best possible alignments early in the procedure, to avoid accumulating errors. To accomplish this, a tree of the sequences is usually constructed to guide the progressive alignment algorithm. And to overcome the problem of a time consuming tree construction step, we are using word matching, a method that group sequences in a very efficient way, saving much time, without reducing the resulting alignment accuracy significantly.
Our algorithm (developed by QIAGEN Aarhus) has two speed settings: "standard" and "fast". The standard method makes a fairly standard progressive alignment using the fast method of generating a guide tree. When aligning two alignments to each other, two matching columns are scored as the average of all the pairwise scores of the residues in the columns. The gap cost is affine, allowing a different cost for the first gapped position and for the consecutive gaps. This ensures that gaps are not spread out too much.
The fast method of alignment uses the same overall method, except that it uses fixpoints in the alignment algorithm based on short subsequences that are identical in the sequences that are being aligned. This allows similar sequences to be aligned much more efficiently, without reducing accuracy very much.