Fastq to Annotated Somatic Variants
The Fastq to Annotated Somatic Variants template workflow identifies somatic variants and annotates these with exon number and amino acid changes.
The workflow can be used to identify and annotate variants in both targeted sequencing and whole genome sequencing pipelines.
The workflow can be found at:
Template Workflows | LightSpeed Workflows (![]() ) | Fastq to Annotated Somatic Variants (
) | Fastq to Annotated Somatic Variants (![]() )
)
If you are connected to a CLC Server via your Workbench, you will be asked where you would like to run the analysis. We recommend that you run the analysis on a CLC Server when possible.
In the first wizard step, select a Reference Data Set (figure 4.10). If you have not downloaded the Reference Data Set yet, the dialog will suggest the relevant data set and offer the opportunity to download it using the Download to Workbench button.
If none of the available reference data sets are appropriate, custom reference data sets can be created, see http://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Custom_Sets.html.
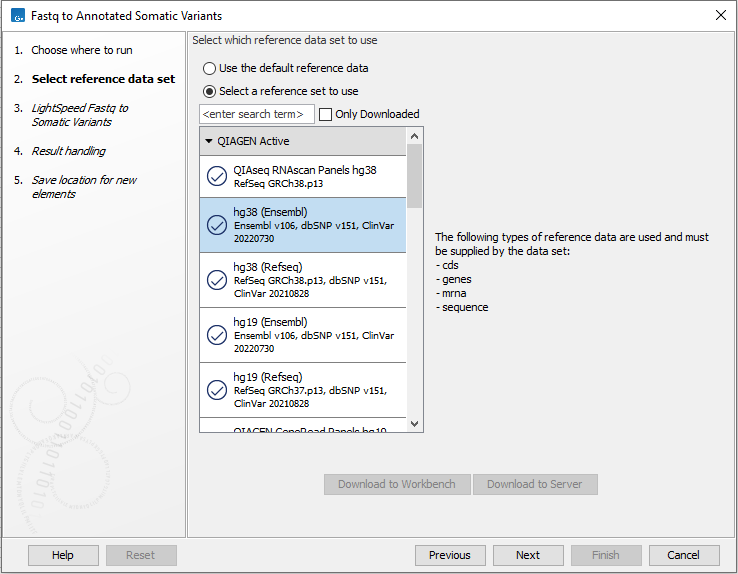
Figure 4.10: Select a reference data set.
In the LightSpeed Fastq to Somatic Variants wizard step (figure 4.11) you have the following options:
- Reads (fastq) Press Browse to select fastq files for analysis.
- Masking mode To enable reference masking when mapping reads, set this option and select a masking track.
- Masking track Provide a masking track for the chosen reference genome if reference masking has been enabled.
- Discard duplicate mapped reads Duplicate mapped reads are per default replaced with a consensus read. Untick if duplicate mapped reads should be retained. See Deduplication for additional details.
- Restrict calling to target regions Optional. If a targeted protocol is used, provide target regions here.
- Lenient inversion detection Enable lenient inversion detection to allow detection of inversions which only has read support in one direction on each of the breakpoints. This is recommended for targeted data. Enabling this option can increase processing time and can result in detection of more false positive inversions.
- Batch Select if fastq files from different samples are used as input, and each sample should be analyzed individually (for information about batching see Batching).
- Join lanes when batching Select to join fastq files from the same sample that were sequenced on different lanes.
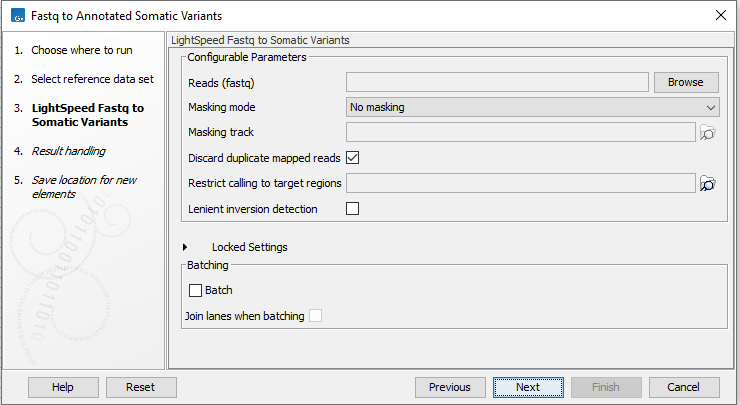
Figure 4.11: Select fastq files.
In the Create Sample Report wizard step, select relevant summary items and specify thresholds for quality control (figure 4.12). Summary items, thresholds and an indication of whether specified thresholds were met, will be shown in the quality control section of the sample report. The default summary items are appropriate for many data sets, but may need to be adjusted.
To add more summary items, press Add..., choose the report type LightSpeed fastq to somatic variants and select summary items as appropriate.
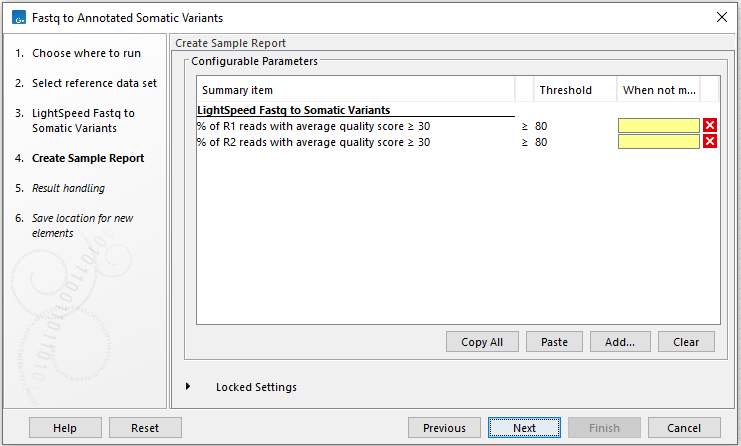
Figure 4.12: Specify summary items. These will be shown in the quality control section of the sample report.
In the final wizard step, choose to Save the results of the workflow and specify a location in the Navigation Area before clicking Finish.
Subsections