Batching
The tools LightSpeed Fastq to Germline Variants, LightSpeed Fastq to Somatic Variants and LightSpeed Fastq to Somatic Variants Tumor Normal have limited batching functionality.
When running the tools from the toolbox, batching is not possible.
LightSpeed Fastq to Germline Variants and LightSpeed Fastq to Somatic Variants
When running the tools as part of a workflow, it is possible to batch over the fastq files (figure 2.2), if not batching over anything else. When batching over the fastq files, the names of the fastq files will determine which fastq files are analyzed together (see Default rules for determining pairs of files here https://resources.qiagenbioinformatics.com/manuals/clcgenomicsworkbench/current/index.php?manual=Illumina.html).
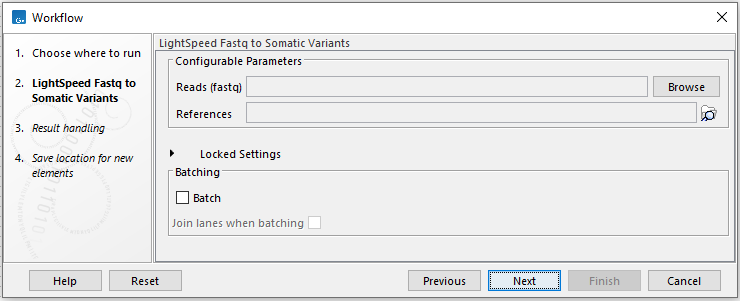
Figure 2.2: When running one of the three LightSpeed Fastq to Variants tools in a workflow, it is possible to batch over the fastq files.
When not batching over fastq files, it is possible to batch over the references, masking track, primers track and target regions tracks, but only if the tracks are added as input elements in a workflow (figure 2.3).
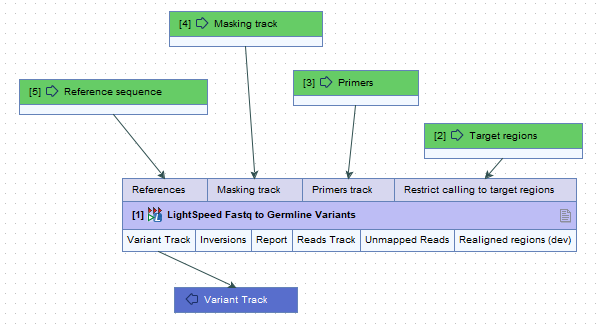
Figure 2.3: When running one of the three LightSpeed Fastq to Variants tools in a workflow, and not batching over fastq files, it is possible to batch over the inputs references, masking track, primers track and target regions when they are added as input elements in a workflow.
LightSpeed Fastq to Somatic Variants Tumor Normal
Only batching over references, masking track, primers track and target regions tracks is possible.