Setting up an experiment
To set up an experiment:
Toolbox | Microarray and Small RNA Analysis (![]() )| Set Up Experiment (
)| Set Up Experiment (![]() )
)
Select the samples that you wish to use by double-clicking or selecting and pressing the Add (![]() ) button (see figure 27.19).
) button (see figure 27.19).
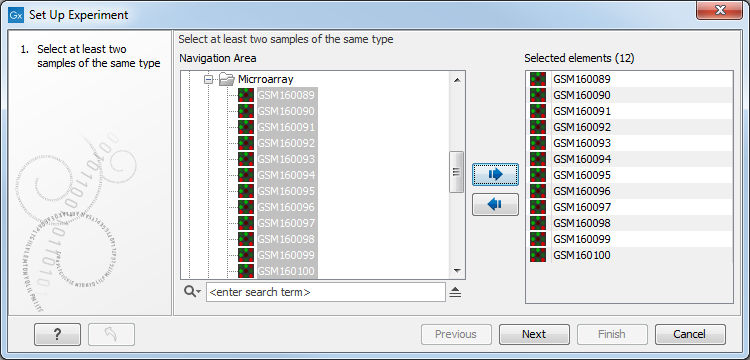
Figure 27.19: Select the samples to use for setting up the experiment.
Note that we use "samples" as the general term for both microarray-based sets of expression values and sequencing-based sets of expression values (e.g. an expression track from RNA-Seq).
Clicking Next shows the dialog in figure 27.20.
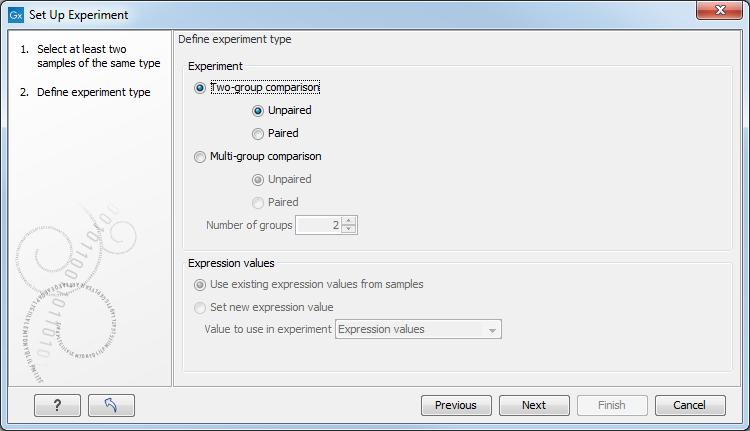
Figure 27.20: Defining the number of groups and expression value type.
Here you define the experiment type and the number of groups in the experiment.
The options are:
- Experiment. At the top you can select a two-group experiment, and below you can select a multi-group experiment and define the number of groups.
Note that you can also specify if the samples are paired. Pairing is relevant if you have samples from the same individual under different conditions, e.g. before and after treatment, or at times 0, 2, and 4 hours after treatment. In this case statistical analysis becomes more efficient if effects of the individuals are taken into account, and comparisons are carried out not simply by considering raw group means but by considering these corrected for effects of the individual. If Paired is selected, a paired rather than a standard t-test will be carried out for two group comparisons. For multiple group comparisons a repeated measures rather than a standard ANOVA will be used.
- Expression values. For RNA-Seq experiments, you can also choose which expression value to be used when setting up the experiment. This value will then be used for all subsequent analyses. If you choose to Set new expression value you can choose between the following options depending on whether you look at the gene or transcript level:
- Genes: Unique exon reads. The number of reads that match uniquely to the exons (including the exon-exon and exon-intron junctions).
- Genes: Unique gene reads. This is the number of reads that match uniquely to the gene.
- Genes: Total exon reads. Number of reads mapped to this gene that fall entirely within an exon or in exon-exon or exon-intron junctions. As for the "Total gene reads" this includes both uniquely mapped reads and reads with multiple matches that were assigned to an exon of this gene.
- Genes: Total gene reads. This is all the reads that are mapped to this gene, i.e., both reads that map uniquely to the gene and reads that matched to more positions in the reference (but fewer than the "Maximum number of hits for a read" parameter) which were assigned to this gene.
- Genes: RPKM. This is the expression value measured in RPKM [Mortazavi et al., 2008]:
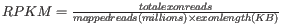 . See exact definition below. Even if you have chosen the RPKM values to be used in the Expression values column, they will also be stored in a separate column. This is useful to store the RPKM if you switch the expression measure.
See more in Definition of RPKM.
. See exact definition below. Even if you have chosen the RPKM values to be used in the Expression values column, they will also be stored in a separate column. This is useful to store the RPKM if you switch the expression measure.
See more in Definition of RPKM.
- Transcripts: Unique transcript reads. This is the number of reads in the mapping for the gene that are uniquely assignable to the transcript. This number is calculated after the reads have been mapped and both single and multi-hit reads from the read mapping may be unique transcript reads.
- Transcripts: Total transcript reads. Once the "Unique transcript read's" have been identified and their counts calculated for each transcript, the remaining (non-unique) transcript reads are assigned randomly to one of the transcripts to which they match. The "Total transcript reads" counts are the total number of reads that are assigned to the transcript once this random assignment has been done. As for the random assignment of reads among genes, the random assignment of reads within a gene but among transcripts, is done proportionally to the "unique transcript counts" normalized by transcript length, that is, using the RPKM. Unique transcript counts of 0 are not replaced by 1 for this proportional assignment of non-unique reads among transcripts.
- Transcripts: RPKM. The RPKM value for the transcript, that is, the number of reads assigned to the transcript divided by the transcript length and normalized by "Mapped reads" (see below).
Clicking Next shows the dialog in figure 27.21.
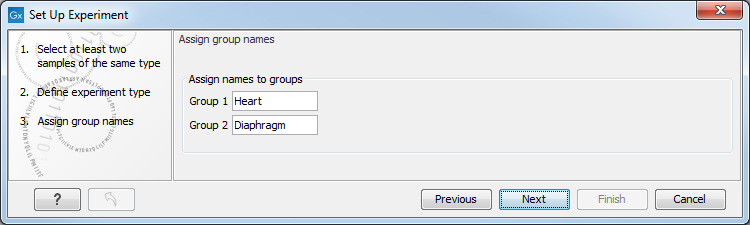
Figure 27.21: Naming the groups.
Depending on the number of groups selected in figure 27.20, you will see a list of groups with text fields where you can enter an appropriate name for that group.
For multi-group experiments, if you find out that you have too many groups, click the Delete (![]() ) button. If you need more groups, simply click Add New Group.
) button. If you need more groups, simply click Add New Group.
Click Next when you have named the groups, and you will see figure 27.22.
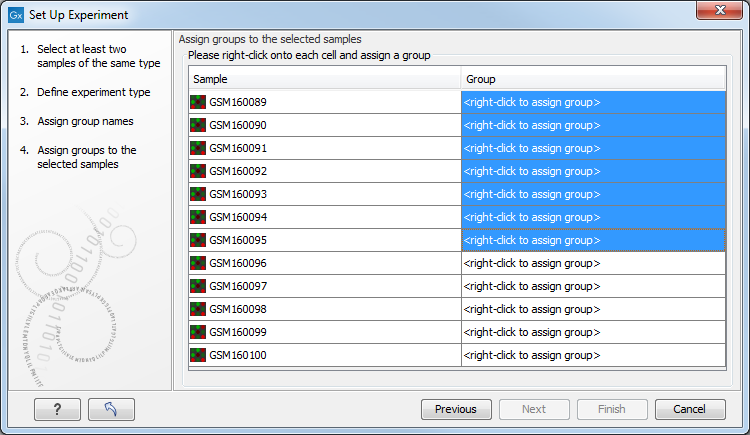
Figure 27.22: Putting the samples into groups.
This is where you define which group the individual sample belongs to. Simply select one or more samples (by clicking and dragging the mouse), right-click (Ctrl-click on Mac) and select the appropriate group.
Note that the samples are sorted alphabetically based on their names.
If you have chosen Paired in figure 27.20, there will be an extra column where you define which samples belong together. Just as when defining the group membership, you select one or more samples, right-click in the pairing column and select a pair.
Click Finish to start the tool.